
Abstract
Liver cancer, primarily hepatocellular carcinoma, represents a major global health issue with significant clinical, economic, and psychological impacts. Its incidence continues to rise, driven by risk factors such as hepatitis B and C infections, nonalcoholic steatohepatitis, and various environmental influences. The Wnt/β-Catenin signaling pathway, frequently dysregulated in HCC, emerges as a promising therapeutic target. Critical genetic alterations, particularly in the CTNNB1 gene, involve mutations at key phosphorylation sites on β-catenin’s N-terminal domain (S33, S37, T41, and S45) and in armadillo repeat domains (K335I and N387 K). These mutations impede β-catenin degradation, enhancing its oncogenic potential. In addition to genetic alterations, molecular and epigenetic mechanisms, including DNA methylation, histone modifications, and noncoding RNAs, further influence β-catenin signaling and tumor progression. However, β-catenin activation alone is insufficient for hepatocarcinogenesis; additional genetic “hits” are required for tumor initiation. Mutations or alterations in genes such as Ras, c-Met, NRF2, and LKB1, when combined with β-catenin activation, significantly contribute to HCC development and progression. Understanding these cooperative mutations provides crucial insights into the disease and reveals potential therapeutic strategies. The complex interplay between genetic variations and the tumor microenvironment, coupled with novel therapeutic approaches targeting the Wnt/β-Catenin pathway, offers promise for improved treatment of HCC. Despite advances, translating preclinical findings into clinical practice remains a challenge. Future research should focus on elucidating how specific β-catenin mutations and additional genetic alterations contribute to HCC pathogenesis, leveraging genetically clengineered mouse models to explore distinct signaling impacts, and identifying downstream targets. Relevant clinical trials will be essential for advancing personalized therapies and enhancing patient outcomes. This review provides a comprehensive analysis of β-Catenin signaling in HCC, highlighting its role in pathogenesis, diagnosis, and therapeutic targeting, and identifies key research directions to improve understanding and clinical outcomes.
Introduction
Globally liver cancer prevalence is ranked sixth, with 865,269 new cases diagnosed annually. It is also the third leading cause of cancer-related mortality, resulting in 757,948 deaths each year. In the United States (US), projections indicate 41,630 new cases and 29,840 deaths from Liver cancer in 2024, making it the 17th most common cause of cancer but the 6th leading cause of cancer-related death. This poses significant clinical, economic, and psychological challenges in developed and developing nations.1-3
The most predominant form of liver cancer is hepatocellular carcinoma (HCC), accounting for approximately 80% of primary liver tumors. 4 Globally and in the US, the incidence of HCC has been rising over the past decade, mainly due to persistent hepatitis B (HBV) and C (HCV) viruses infections, alcoholic liver disease as well as the increasing prevalence of metabolic dysfunction associated steatohepatitis (MASH) since 2000. 5
Hepatocarcinogenesis unfolds through several stages, starting most frequently with the forming of precancerous lesions such as dysplastic nodules within cirrhotic livers and rarely through neoplastic transformation of hepatocellular adenomas within healthy liver tissue. 6 The interplay between pathways that regulate metabolism, apoptosis, chromatin biology, splicing, proliferation, and cell cycle significantly contribute to HCC development. 7 Among these, the Wnt/β-Catenin signaling pathway is critical in tumor growth, apoptosis, retention, and migration. 8 Hyperactivation of this pathway occurs in approximately one-third of all HCC cases and is a principal or truncal oncogenic driver. 9
The Wnt gene family, first identified in the late 20th century through studies in Drosophila and mice, combines the name of the “wingless” gene involved in embryonic development and the “int-1” proto-oncogene associated with cancer. Recognizing their critical roles, researchers have since discovered over 100 homologous Wnt genes across various eukaryotic species, highlighting the evolutionary conservation and significance of Wnt signaling in development and disease. 10 A central component of the Wnt signaling pathway is β-catenin, independently discovered in the late 1980s due to its dual roles in cell adhesion and signaling. Initially identified as a structural protein that links E-cadherin to the cytoskeleton, β-catenin was also found to be a critical player in Wnt signaling, as demonstrated by its Drosophila counterpart, Armadillo. Further research revealed that β-catenin is essential not only for maintaining cellular architecture but also for regulating gene expression during development. 11 Moreover, β-catenin has been linked to various oncogenic processes. 12
In the early 2000s, mouse model studies provided significant insights into the role of β-catenin in hepatocarcinogenesis. For instance, research by Aydinlik et al. showed that the mutational activation of the Wnt/β-catenin signaling pathway could drive liver tumor development in mice. In this study, six-week-old mice received a single dose of the liver carcinogen N-nitrosodiethylamine (DEN), followed by chronic treatment with the tumor promoter phenobarbital (PB). This initiation/promotion regimen resulted in the preferential growth of hepatoma cells with CTNNB1 mutations which is the mouse equivalent of the human CTNNB1 gene. Remarkably, approximately 80% of the liver tumors that developed harbored mutations in CTNNB1. 13
This laid the ground over past decades for numerous studies which have evaluated genetic profiles linked to cancer susceptibility, revealing a notable prevalence of β-Catenin mutations in HCC.14-16 The CTNNB1 gene, which encodes β-catenin, is identified as the most commonly mutated oncogene in HCC, with mutation rates reported between 16% and 40% across various studies.17-21 Additionally, loss of function mutations in gene encoding Axin1, a scaffolding protein essential for β-catenin’s degradation, can occur in an additional 11% of HCC. However, these mutations surprisingly have only a minor impact on the activation of Wnt pathway target genes, suggesting their primary effects may involve non-Wnt related pathways, which remain to be fully explored.21,22 Finally mutation of axin 2 which has a role similar to axin 1 is seen in an additional 3% of cases. 14
Unraveling the β-Catenin pathway promises to identify therapeutic targets that can significantly improve patient outcomes.
In this review, we delve into the role of β-Catenin in HCC. We start with a thorough overview of HCC’s epidemiology, prevalence, and biology, including its molecular subtypes. We then examine β-Catenin mutations, their interactions with other oncogenes and tumor suppressor genes, and associated clinical risk factors. The review also covers tumor classification and pathological features specific to β-Catenin mutant HCC, interactions within the tumor microenvironment, and the epigenetic and genetic variations impacting β-Catenin mutations. Finally, we evaluate various strategies for targeting the β-Catenin pathway and discuss its potential as a diagnostic tool and prognostic marker in clinical settings.
Epidemiology, Clinical Significance, and Molecular Mechanisms of β-Catenin Mutations in Hepatocellular Carcinoma
Primary liver cancer begins in the liver and encompasses several subtypes, such as hepatocellular carcinoma, intrahepatic cholangiocarcinoma, angiosarcoma, hemangiosarcoma, and hepatoblastoma. 23 Based on the most recent GLOBOCAN estimates from the International Agency for Research on Cancer (IARC) statistics, primary liver cancer stands as the third leading cause of cancer-related death and sixth most common cancer worldwide. 3 HCC predominates as the primary type of liver cancer worldwide, accounting for nearly 75 percent of all cases. 24
Epidemiological studies identified HBV as a primary causative agent of liver cancer, although other etiological factors have also been recognized. 25 Asia and Africa have the highest incidence rate. 26 While incidence rates of HCC have declined in certain high-rate regions, they have risen in many low-incidence areas. 27 Between 1978 and 2012, HCC incidence decreased in Italy and several Asian countries but surged in India, Oceania, the Americas, and most European countries. However, the increasing trend in some nations, such as the United States, has slowed down, with rates in various subgroups stabilizing or declining in recent years. 28
The prognosis for HCC remains poor across all regions worldwide, leading to roughly equivalent incidence and mortality rates. In 2022 out of every 100,000 people, 10.9 were diagnosed with liver cancer, and 9.5 died from it. 3
Despite significant advances, numerous molecular mechanisms involved in HCC tumor progression remain elusive. 25 The β-Catenin protein is crucial in this context, as it supports cell adhesion and transmits proliferative signals within the Wingless/Wnt pathway. 29 Dysregulation of this pathway plays a pivotal role in carcinogenesis.
Typically, β-Catenin is located at the lateral cell membrane as part of the E-cadherin-catenin complex, part of the Adherens junctions (AJ). This complex is seen in hepatocytes and is present all across the liver lobule. Intriguingly, studies in genetic mouse models have shown that loss of β-catenin at AJ can be compensated by upregulation of γ-catenin, which is usually a desmosomal protein. 30 In fact, γ-catenin bound to E-cadherin to maintain AJ when β-catenin was conditionally deleted from hepatocytes.31,32 Additionally, β-catenin resides in the cytoplasm and nucleus in the hepatocytes in zone-3 or pericentral region of the liver lobule, where it acts as a mediator of Wingless/Wnt-dependent signal transduction.33-36 Here it controls expression of specific genes and thus essential for the process of metabolic zonation.
As the pivotal component of the common Wnt signaling pathway, β-Catenin possesses multiple biological functions, including oncogenic ones. Numerous studies have demonstrated genetic profiles over the past decades associated with cancer predisposition, consistently identifying a high frequency of β-Catenin mutations in HCC.14,16,37
The CTNNB1 gene, which encodes β-catenin, is identified as the most frequently mutated oncogene in HCC, with mutation rates reported between 16% and 40% across various studies.17-21
The Wnt/β-catenin signaling pathway is initiated when Wnt proteins, which are secreted and undergo post-translational modifications such as lipid modification by the enzyme PORCN, bind to receptors on the cell surface. The main receptors involved are Frizzled (FZD) and low-density-lipoprotein-receptor-related-protein 5/6 (LRP5/6), which work together to activate the pathway. Upon Wnt binding, the protein Dishevelled (DVL) is recruited to the plasma membrane, where it facilitates the formation of a signaling complex by clustering FZD and LRP6 and promoting LRP6 phosphorylation. This process inhibits the β-catenin destruction complex, a group of proteins including AXIN, APC, GSK-3β, and CK-1α that normally work together to target β-catenin for degradation. 38
In the absence of Wnt signals, this destruction complex phosphorylates β-catenin through the E3 ubiquitin ligase β-transducin repeat containing protein (E3 ligase β-TRCP), marking it for degradation by the proteasome. However, when Wnt signaling is activated, the destruction complex is disrupted, allowing β-catenin to stabilize and accumulate in the cytoplasm. β-catenin then translocates to the nucleus, where it interacts with TCF/LEF transcription factors to regulate the expression of Wnt target genes namely MYC, CYCLIN D1,OAT, GLUL, AXIN2, TBX3, SP5, and LGR5 involved in cell proliferation and survival.39,40 This pathway is critical for development and tissue homeostasis, and its dysregulation is implicated in various cancers, including HCC. 38
Figure 1 gives an overview of this process and how it leads to the development of HCC. CTNNB1 Mutation and canonical wnt pathway in HCC.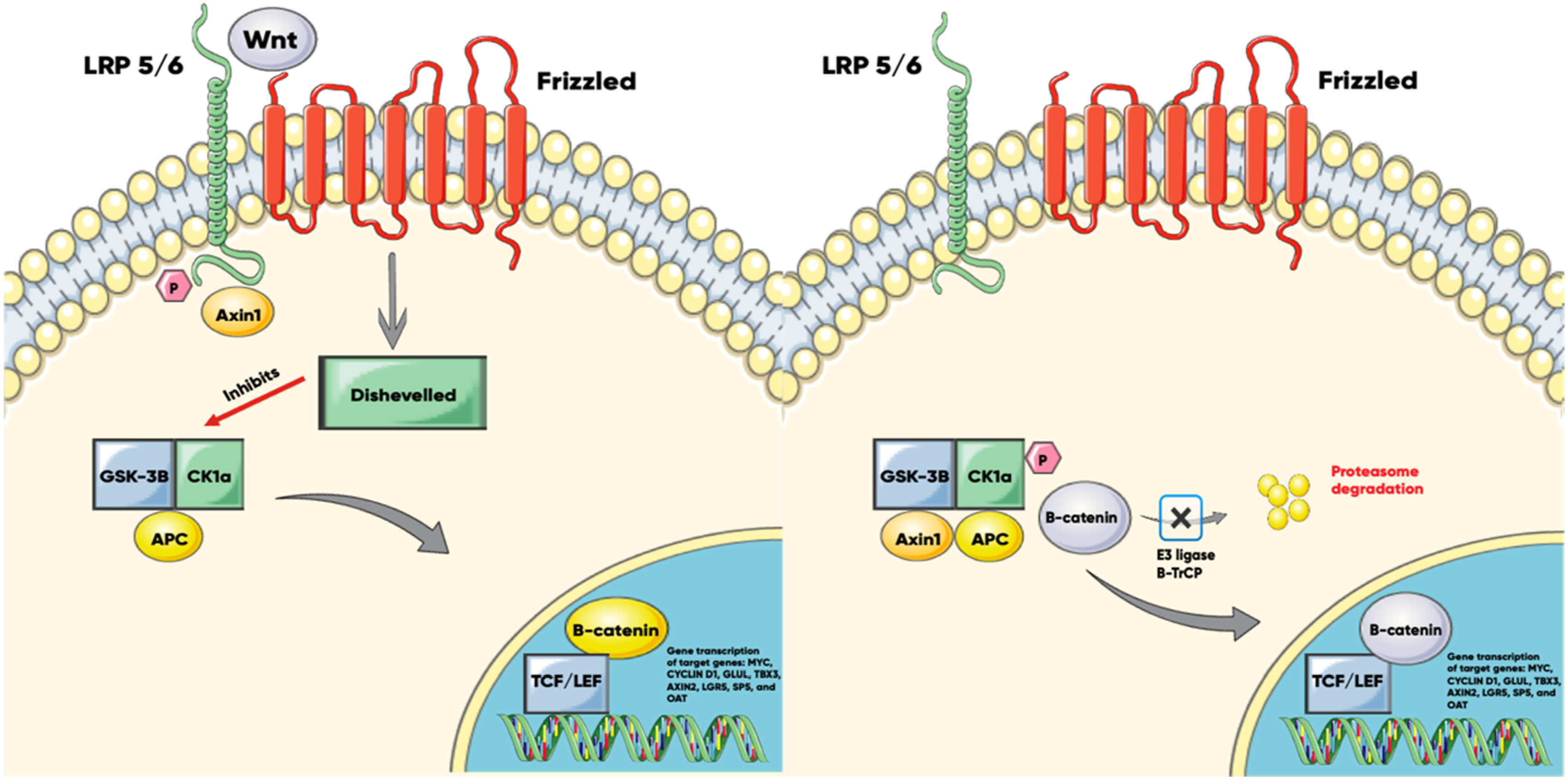
Left (Activation of Wnt Signaling)
When Wnt signaling is activated, Dishevelled acts as a mediator between the destruction complex and the cell surface receptors. It bridges the communication by interacting with Frizzled and Axin1, which binds to the phosphorylated LRP5/6. This interaction disrupts the destruction complex, allowing β-catenin to stabilize and translocate to the nucleus. Inside the nucleus, β-catenin binds to TCF/LEF transcription factors to regulate the expression of target genes, including MYC, CYCLIN D1, OAT, GLUL, AXIN2, TBX3, SP5, and LGR5.
Right (Inactivation of Wnt Signaling and Mutated β-Catenin)
In the absence of a Wnt signal, the scaffold proteins APC and Axin1 bind β-catenin, recruiting the kinases CK1α and GSK3β. CK1α initiates a priming phosphorylation on β-catenin, which is further phosphorylated by GSK3β. The phosphorylated β-catenin is then recognized and ubiquitinated by the E3 ligase β-TrCP, marking it for degradation by the proteasome. However, in cases where the CTNNB1 gene is mutated, the gain-of-function mutation prevents the phosphorylation and subsequent degradation of the β-catenin protein. This leads to its stabilization and nuclear translocation, contributing to the oncogenic process in HCC by continuously transcribing the target genes.
GSK-3β glycogen synthase kinase-3, AXIN axis inhibition protein, CK1 casein kinase 1, APC adenomatous polyposis coli, TCF T cell factor, LEF lymphocyte enhancer factor-1, LRP low-density-lipoprotein-receptor-related-protein, β-TrCP Beta-transducin repeats-containing proteins, OAT Ornithine Aminotransferase, GLUL Glutamate-Ammonia Ligase, TBX3 T-box 3, LGR5 Leucine Rich Repeat Containing G Protein-Coupled Receptor 5.
β-catenin Mutations and Crosstalk With Other Oncogenes and Tumor Suppressor Genes in HCC
Exon 3 of the CTNNB1 gene is crucial for the degradation of β-catenin, as it encodes key phosphorylation sites on its N-terminal domain for serine and threonine residues. These include S33, S37, and T41, which are phosphorylated by GSK-3β, triggering the proteasomal degradation of β-catenin. S45 is targeted by casein kinase-1, while D32 and G34 are essential for the interaction of β-catenin with Fbw1, a part of the E3 ligase complex β-TRCP. Most CTNNB1 mutations occur at these critical sites—S33, S37, S45, T41, D32, and G34—resulting primarily in missense mutations that substitute one amino acid for another. 41 Additionally, recent studies have identified missense mutations in the armadillo repeat domains 5 and 6 of β-catenin, specifically K335I and N387 K, which, in conjunction with MET, promoted liver tumor formation in mice. 42 The precise mechanism of oncogenesis for these mutations remains unclear, but research by Liu et al. suggests it may involve decreased affinity for APC while preserving TCF/LEF binding. 42
Despite these findings, β-catenin activation alone in animal studies is not sufficient to induce spontaneous liver tumors due to its role in a p53-dependent DNA damage response and subsequent cell senescence, which impede tumor growth and oncogenesis. 43 studies using an inducer/promoter regimen to generate CTNNB1-mutated liver tumors in mice demonstrated that phenobarbital (PB) promotes these tumors. 13 PB is a xenobiotic and a known activator of Constitutive Androstane Receptor (CAR), which regulates genes involved in xenobiotic metabolism and detoxification in the liver. 44 Research by Dong et al. showed that CAR activation helps overcome the senescence induced by β-catenin activation, providing one of the additional “hits” necessary for oncogenesis. 43
The ras family genes encode small GTPase proteins that relay signals from transmembrane receptors to the nucleus. Mutations in Ras disrupt GTPase activity and lead to continuous activation of downstream signaling pathways. 45 Harada et al. created an innovative mouse model of HCC by incorporating β-catenin and H-Ras mutations through an adenovirus-mediated liver-specific Cre-expression system. Their study demonstrated that while β-catenin or H-Ras mutations individually did not induce HCC, the combination of these mutations led to tumor formation in every mouse tested. This observation aligns with human HCC, where β-catenin mutations, H-Ras mutations, and elevated levels of transforming growth factor β (TGF-β) or insulin-like growth factor II (IGF-II) associated with increased Ras signaling are commonly observed. The model underscored those simultaneous mutations prompt rapid liver tumor development, highlighting the essential role of these genetic alterations in driving hepatocarcinogenesis. 46
c-Met is a receptor tyrosine kinase from the MET family, expressed on the surface of various cell types. Its ligand, hepatocyte growth factor (HGF), binds to c-Met and initiates signaling pathways crucial for normal processes like embryogenesis and wound healing. In cancer, abnormal activation of the HGF/c-Met axis, often linked to mutations or overexpression of the c-Met gene, drives tumor growth and progression. 47 Tward et al. conducted a study that revealed tumorigenesis in the liver initiated by MET can follow different paths depending on the sequence of genetic alterations. If β-catenin activation occurs first, HCC develops; however, MET inactivation in this context prevents tumor formation by β-catenin alone. Alternatively, when MET mutation is followed by HNF1α inactivation, the pathway leads to hepatic adenoma (HCA). 48
LKB1 is a serine/threonine kinase that phosphorylates and activates several kinases, including AMP-activated protein kinase (AMPK) and microtubule-associated protein/microtubule affinity-regulating kinases (MARK). In a study led by Miyoshi et al., the loss of LKB1 in hepatocytes was found to trigger the formation of multiple hepatic nodular foci, with subsequent activation of β-catenin driving the progression of these foci into HCC. 49
NRF2 is a transcription factor that plays a key role in cellular defense against oxidative stress by regulating the expression of antioxidant genes. Tao et al. demonstrated that activation of the Nrf2 signaling pathway in HCC can result from gain-of-function mutations in NFE2L2 or loss-of-function mutations in KEAP1, both of which disrupt the degradation of NRF2. By co-expressing these NFE2L2 mutations with β-catenin mutations in mouse hepatocytes using the sleeping beauty transposon/transposase system, they successfully induced HCC. The resulting tumors exhibited characteristics similar to a subset of human HCCs with activated Nrf2 and β-catenin pathways, and they were responsive to mTOR inhibitors. 50
In summary, β-catenin activation alone is insufficient to drive hepatocarcinogenesis, requiring additional genetic “hits” to initiate tumor formation. Various studies have shown that these additional hits can come from mutations or alterations in Ras, c-Met, NRF2 or LKB1, which, when combined with β-catenin activation, significantly contribute to the development and progression of hepatocellular carcinoma. Understanding these cooperative mutations provides a significant tool for understanding the disease and exploring therapeutic options.
Clinical Risk Factors for HCC and β-Catenin Mutation
Hepatitis B and C infections account for about 75% of HCC cases, with the remaining instances linked to genetic disorders, environmental toxins such as aflatoxin, dietary factors including alcohol use, and metabolic syndrome. 51 Each of these pathways involves distinct molecular alterations that contribute to the development of HCC, their relationship with the CTNNB1 mutations is presented below.
Fatty liver disease can result from various factors, including alcohol abuse and metabolic syndrome. Although the progression from steatosis to HCC is well-documented, the specific mechanisms remain partially understood. 52 Stauffer et al. provided new insights by creating a novel mouse model to investigate how lipogenic traits, often linked with diet-induced metabolic disorders, contribute to liver cancer. Their study used the Sleeping Beauty transposon system to co-express AKT and β-catenin genes, leading to the development of tumor foci within 4 weeks. Tumors initially appeared as steatotic hepatocellular adenomas and progressed to HCC after in vivo passage. Molecular analysis of these tumors showed they align with specific human HCC subtypes associated with poor outcomes, emphasizing the involvement of steatosis and lipid metabolism pathways. This model highlights the role of AKT and β-catenin coactivation in simulating a lipogenic tumor phenotype and offers a useful approach for investigating the transformation from steatotic lesions to carcinoma. 53
HCCs linked to hepatitis B virus (HBV) infection are most prevalent in Southeast Asia and sub-Saharan Africa, while those associated with hepatitis C virus (HCV) are more common in southern Europe and Japan. In countries like Italy, Spain, and Japan, the majority of HCC cases are related to HCV infection. 54 Research suggests that β-catenin overexpression and mutations are more commonly observed in HCV-related HCCs. A study by Huang et al. found β-catenin mutations in 41% of HCV-related HCCs cases, compared to only 18% in HBV-related HCCs, as reported by Javanmard et al. Furthermore, β-catenin mutations in HCV-associated HCCs often occur in noncirrhotic livers, lacking the typical risk factors commonly seen in HBV-related HCCs.20,54,55 The mechanisms of oncogenesis differ between HBV and HCV. In chronic HBV infection, viral DNA integration and the hepatitis B X protein play critical roles in driving cancer development. In contrast, HCV promotes oncogenesis by activating multiple oncogenic pathways, either through autocrine signaling or phosphorylation of intracellular messengers. 56 Telomeres, the DNA-protein structures at the ends of chromosomes, are crucial for cell longevity, and their maintenance is often linked to cancer. Increased activity of telomerase reverse transcriptase (TERT) is found in many cancers, including HCC. Several studies have shown that TERT and CTNNB1 mutations are more prevalent in HCV-related HCCs than in HBV-related cases.57,58 Notably, research by Pezzuto et al. revealed that these mutations co-exist in over half of the cases, suggesting a significant interaction between these genetic alterations in the development of virus-induced HCC. 57
Alcohol and aflatoxin B1 (AFB1) are significant contributors to a subset of HCC cases. The progression from alcoholic fatty liver disease (AFLD) to alcoholic steatohepatitis (ASH), cirrhosis, and ultimately HCC is well-established. This progression is largely driven by oxidative stress induced by acetaldehyde, a toxic metabolite of alcohol. 56 An exome sequencing analysis of 243 liver tumors revealed a strong association between alcohol-related HCC and CTNNB1 mutations. 59 Conversely, AFB1 is a potent carcinogen that promotes HCC by forming DNA adducts through its reactive intermediate, AFB1-8,9-epoxide (AFBO), which induces genetic alterations in liver cells, most notably mutations in the p53 gene. HCC is particularly prevalent in regions with high AFB1 exposure levels. 60 In a study of HCC samples from individuals with significant AFB1 exposure in Guangxi, China, CTNNB1 mutations were identified in only 5 out of 62 cases (approximately 8%), including four-point mutations and one deletion in the phosphorylation region of codons 32-45, which are critical for β-catenin regulation. Despite the low frequency of CTNNB1 mutations, immunohistochemical analysis showed that 20 out of 41 HCC samples exhibited positive β-catenin staining, indicating protein accumulation. This suggests that while AFB1 exposure may not frequently cause CTNNB1 mutations, it could still lead to β-catenin accumulation and possibly activate Wnt signaling pathways. These findings imply that other components of the Wnt pathway, such as axin or APC, may also contribute to AFB1-related hepatocarcinogenesis, highlighting the complex molecular mechanisms driving liver cancer associated with aflatoxin exposure. 61
In conclusion, the development of HCC is driven by a variety of etiological factors, each involving distinct molecular pathways. Hepatitis B and C infections account for the majority of HCC cases worldwide, with varying regional prevalence. HCV-related HCCs show a higher frequency of β-catenin mutations compared to HBV-related cases. Genetic mutations, environmental toxins like aflatoxin B1, alcohol abuse, and metabolic conditions further contribute to HCC development through diverse mechanisms, including oxidative stress, DNA adduct formation, and disruption of cellular signaling pathways. it is crucial to recognize the limitations of animal models but the interplay between these factors and β-catenin mutations underscores the complexity of hepatocarcinogenesis and highlights the need for further research into the specific molecular alterations associated with different HCC risk factors.
Tumor Microenvironment interactions in β-Catenin Mutant HCC
A tumor is a complex tissue composed of cancerous cells, stromal cells, inflammatory cells, blood vessels, and extracellular matrices (ECM), collectively referred to as the tumor microenvironment (TME). 62 The composition of the TME gives rise to different immunological phenotypes, or ‘immunotypes,' which influence the tumor’s prognosis and response to immune checkpoint inhibitors (ICIs): immune-inflamed (best prognosis), immune-excluded, and immune-desert (worst prognosis). The TME’s components can exhibit pro-cancer, anti-cancer, or dual effects. Key players include myeloid cells, cancer-associated fibroblasts (CAFs), tertiary lymphoid structures (TLS), and various molecules secreted within the TME. 63 A thorough understanding of the interactions among these elements is crucial for developing effective treatment strategies.62,63 Here we are going to discuss the link between the TME and the Wnt/β-Catenin pathway in HCC.
Tumor-associated macrophages (TAMs), a subtype of myeloid cells within the TME, have dual origins: some arise from tissue-resident macrophages with an embryonic origin, while others are derived from circulating monocytes that originate from hematopoietic stem cells (HSCs). 64 TAMs can adopt different phenotypes, with the two main types being M1 and M2. M1 macrophages, induced by IFN-γ and LPS, exhibit anti-tumor properties, whereas M2 macrophages, stimulated by IL-4 and IL-13, are associated with pro-tumor activities such as promoting cell proliferation, angiogenesis, metastasis, and epithelial-to-mesenchymal transition (EMT) through the secretion of cytokines, growth factors, chemokines, and matrix metalloproteinases (MMPs).65,66 In the liver, Kupffer cells (KCs) serve as the primary TAMs in HCC. 65
During chronic liver disease, hepatic macrophages release Wnt-3a following the phagocytosis of hepatocyte debris. Wnt-3a activates the Wnt/β-Catenin signaling pathway in hepatic progenitor cells (HPCs), promoting their differentiation into hepatocytes while inhibiting cholangiocyte specification by counteracting Notch signaling. 67 However, inappropriate activation of the Wnt/β-Catenin pathway is implicated in carcinogenesis. A study by Yang et al. found that Wnt signaling from HCC tumors polarizes TAMs toward the M2 phenotype, as indicated by the upregulation of target genes such as c-Myc, cyclin D1, and Axin2. This polarization was blocked when Wnt genes were knocked down in HCC cells. Additionally, the study suggested that this M2 polarization, which has pro-tumor properties, could occur in an autocrine manner by macrophages themselves, as observed in vivo. 68
In both mouse models and human HCC, β-catenin activation, especially through CTNNB1 mutations, has been strongly associated with immune exclusion. This exclusion is thought to occur due to reduced recruitment of CD103+ dendritic cells, driven by lower production of chemokines like CCL5, which usually attract T-cells to the tumor microenvironment. Additionally, there is an increase in pro-tumor M2 macrophages, contributing to the immune-excluded phenotype. Lower PD-L1 expression has also been observed in these cases, which may explain the reduced effectiveness of immune checkpoint inhibitors (ICIs). Studies consistently show that HCCs with CTNNB1 mutations are linked to the immunotype with the poorest prognosis. These mutations have emerged as significant biomarkers for poor immunotherapy response due to their strong association with immune exclusion.69-71
Cancer stem cells (CSCs) are a distinct subset of undifferentiated cells characterized by their abilities for self-renewal and pluripotency, mirroring the functions of typical somatic stem cells (SSCs). Initially identified in human acute myeloid leukemia (AML) through their high tumorigenic potential in mouse models, CSCs have been recognized as critical drivers of cancer progression. Two primary models explain CSC behavior: the classical unidirectional differentiation model, where CSCs differentiate into non-CSCs, and the plastic bidirectional model, which allows non-CSCs to revert back into CSCs. These dynamic processes are tightly regulated by various signaling pathways, notably the Wnt/β-catenin pathway. 72
Aberrant activation of Wnt signaling plays a pivotal role in the maintenance and regulation of CSCs across multiple cancer types. It promotes CSC persistence by enabling non-CSCs to acquire stem-like properties through mechanisms such as epithelial-mesenchymal transition (EMT) and supports the persistence of the CSC population. 72 Recent studies have shown that CSCs within the TME are linked to aggressive tumor progression, resistance to chemotherapy which often target rapidly dividing cells but may not effectively eliminate CSCs, and recurrence in HCC patients. 72 Markers such as epithelial cell adhesion molecule (EpCAM), CD44, LGR5, and CD133 which are downstream targets of the Wnt/β-catenin signaling pathway, have been identified as prevalent CSC indicators in HCC.73-75
Supporting this association, a study by Pandit et al. demonstrated increased expression of CSC markers and elevated β-catenin levels in Hepa 1-6 spheroids within an orthotopic HCC mouse model. Notably, the knockdown of the CTNNB1 gene using siRNA led to a loss of the spheroid phenotype and induced differentiation, underscoring the integral role of β-catenin in the formation and maintenance of CSCs in HCC. 76 Furthermore, it was shown by Khosla et al. that there is an increased secretion of wnt3 in an autocrine manner by EpCam + CSCs in patients with advanced cirrhosis that developed HCC further acknowledging the role of these cell in HCC development. 77
Vasculogenesis refers to the de novo formation of blood vessels, while angiogenesis is the process by which new blood vessels develop from pre-existing ones. Tumor angiogenesis is a critical factor in tumor growth, progression, and metastasis. When the balance between proangiogenic factors and those that inhibit angiogenesis shifts toward the former, the process of angiogenesis is initiated. 78 A study by Guo et al. found that Ribosomal Protein S15 A (RPS15 A) is overexpressed in HCC and is associated with poor patient survival and increased microvascular density, indicating enhanced angiogenesis. RPS15 A was shown to boost angiogenesis by enhancing Wnt/β-catenin signaling, leading to increased FGF18 expression promoting tumor growth. 79
ECM in tumors consists of various molecules like collagen, elastin, and proteoglycans, primarily produced by CAFs and tumor cells. 80 In HCC, the cysteine-rich protein 61 (Cyr61), a member of the CCN family (which includes CYR61, CTGF, and NOV, respectively known as CCN1, CCN2, and CCN3), is upregulated via the β-catenin pathway. This upregulation, as shown by Li et al., enhances hepatic stellate cell function, promoting cancer progression.81-83 Additionally, Liu et al. demonstrated that knocking down FBXO17, a negative regulator of GSK-3β, led to reduced expression of key proteins in the Wnt/β-catenin pathway, including β-catenin, c-Myc, cyclin D1, MMP-9, and MMP-2, while increasing GSK-3β levels, suggesting FBXO17’s involvement in HCC through this pathway. 84 Furthermore, Shang et al. found that sulfotransferase 2B1b (SULT2B1b) promotes EMT and enhances tumor migration and invasion via the β-catenin/MMP7 pathway. 85 In another study by Cui et al., upregulated FGF15/FGFR4 signaling in mouse models was shown to drive HCC development by activating EMT and Wnt/β-catenin signaling in a lipid metabolic disorder microenvironment. The human homolog, FGF19, is expressed in HCC but not in normal liver cells, suggesting its potential role in HCC progression. 86
In summary, TME plays a pivotal role in shaping the behavior and progression of HCC through intricate interactions between cancer cells, stromal components, and signaling pathways. The Wnt/β-catenin pathway emerges as a key regulator within this dynamic, influencing immune evasion, cancer stem cell maintenance, and angiogenesis. Studies have demonstrated that aberrant activation of this pathway not only promotes the progression of HCC but also contributes to resistance against therapies, including immune checkpoint inhibitors. Although much of the current understanding is based on mouse studies, these findings help pave the way for developing more effective therapeutic strategies targeting the TME and its associated signaling pathways in HCC.
Tumor Classification and Pathological Features of β-catenin Mutant HCC
In 2007, Boyault et al. conducted a transcriptome analysis of 120 HCC tumors, classifying them into six subgroups: G1, G2, G3, G4, G5, and G6. The G1 group primarily included HBV-related tumors in young African patients, often with frequent AXIN1 mutations. The G6 group consisted exclusively of tumors with CTNNB1 mutations, while the G5 group also had a high incidence of CTNNB1 mutations. Notably, AXIN1-mutated tumors were found in different groups than those with CTNNB1 mutations, suggesting distinct molecular mechanisms underlying HCC development. Groups G2, G3, and G4 were characterized by other mutations, including PIK3CA, TP53, CDKN2A, and TCF1. G1 to G3 tumors exhibited chromosomal instability, whereas G4 to G6 tumors were chromosomally stable. This classification provides a strong foundation for further exploration of the molecular diversity in HCC. 87
Another molecular classification was attempted by Hoshida et al., who identified three subclasses of HCC: S1, S2, and S3. These subclasses correlate with clinical parameters such as tumor size, cellular differentiation, and serum alpha-fetoprotein levels. The S1 subclass is notable for abnormal activation of the Wnt signaling pathway, not through CTNNB1 mutations as expected, but via activation of the transforming growth factor β (TGF-β) gene. This leads to a more invasive and aggressive tumor phenotype, characterized by early recurrence, increased vascular invasion, and satellite lesions. These findings highlight that Wnt pathway activation can occur without β-catenin mutations, opening new possibilities for targeted therapies. In contrast, the S3 subclass, where CTNNB1 mutations were more common, showed better differentiation, smaller tumor size, and improved survival outcomes. 88
HCCs with β-Catenin mutations not only promote carcinogenesis but also accelerate bile production. These HCCs may represent a distinct subtype characterized by specific pathological and clinical features, making the radiomics models for predicting HCCs β-Catenin mutation status particularly significant in routine clinical practice in the future.88,89
HCCs with less aggressive pathological features that harbor β-catenin mutations display distinct imaging traits. These include increased enhancement ratios, resulting in a higher contrast-to-noise ratio, during the hepatobiliary phase of gadoxetic acid-enhanced MRI, higher apparent diffusion coefficients (ADC) and a lower contrast-to-noise ratio at diffusion-weighted imaging (DWI) indicating free diffusion. Imaging characteristics, particularly during the hepatobiliary phase, could prove helpful in identifying HCCs that harbor β-Catenin mutation. 90 Recently, AI-based algorithms analyzing MRI and CT scans of HCCs have enabled the radiomics-based diagnosis of β-catenin mutations in HCCs. By integrating LI-RADS descriptors especially the “nodule-in-nodule appearance” with radiomics features derived from CT and MRI, β-catenin activation status in HCC has been detected confidently with an AUC ranging from 0.8 to 0.9.
That being said this study unlike the one previously mentioned did not find any statistically significant difference on the DWI sequence between β-catenin mutated and wild type. Nevertheless, this advancement paves the way for precision medicine approaches in managing HCC. 91
As previously described activation of the Wnt/β-Catenin pathway results in the accumulation of β-Catenin in the cytoplasm and its subsequent transfer into the nucleus, thereby stimulating the transcription of target genes. Detecting nuclear β-Catenin expression through immunohistochemical studies confirms Wnt/β-Catenin pathway activation. 92 However, the reliability of immunohistochemical findings for nuclear β-Catenin expression is limited in terms of sensitivity and specificity. Only 63% of HCCs confirmed by polymerase chain reaction to harbor β-Catenin mutations displayed nuclear or cytoplasmic β-Catenin expression in immunohistochemical analysis. 90 Glutamine synthetase (GS), a downstream target of β-catenin, is a more faithful biomarker of β-catenin mutations and activity of the Wnt pathway both in hepatocellular adenomas as well as HCCs.93,94 Staining for GS has been proposed as a marker of Wnt pathway activation in HCC, although results are varied across centers.95-97
A study by Chiu et al. demonstrated that the inhibition of glutamine synthesis using Crisantaptase and methionine L-sulfoximine significantly reduced the growth of CTNNB1-mutated cells both in vitro and in vivo, highlighting the dependency of these HCC cells on the glutamine pathway. 98 Furthermore, GS-mediated glutamine synthesis is believed to enhance the activity of mammalian target of rapamycin complex 1 (mTORC1) through phosphorylation of mTOR at Ser2448, forming a β-catenin–GS–mTORC1 axis that presents a potential therapeutic target. Additionally, CTNNB1-mutated HCC cells are often positive for p-mTOR-S2448, suggesting that this could serve as an additional marker for these tumors. 97
Overall, different molecular classifications of HCC have emerged, offering insights into tumor heterogeneity and guiding targeted therapies. For instance, classifications based on transcriptome analysis have identified subgroups with distinct molecular and clinical features, such as β-catenin mutations and other specific markers. These classifications provide a robust foundation for understanding HCC diversity and improving treatment strategies.
Immunohistochemistry remains the standard method for assessing mutation status in HCC, providing reliable data on the presence and activity of mutations like β-catenin. This technique is pivotal for guiding diagnosis and treatment. However, advances in radiomics offer a promising complementary approach. Radiomics, which involves extracting and analyzing quantitative features from medical images, has the potential to non-invasively determine HCC mutation status, including β-catenin mutations. Although still evolving, radiomics could enhance our ability to characterize tumors and tailor therapies more precisely, paving the way for more personalized treatment strategies while continuing to support and refine traditional methods like immunohistochemistry.
Molecular Epigenetic Mechanisms Related to β-Catenin Regulation in HCC
Multiple epigenetic dysregulations contribute to the activation of the Wnt/β-Catenin pathway in HCC.
DNA Methylation
DNA methylation, particularly at CpG islands within promoter regions, plays a critical role in regulating gene expression, which is essential for maintaining CSCs properties. 99
This epigenetic modification impacts the Wnt/β-catenin pathway, a process that can be investigated using techniques such as reverse transcriptase polymerase chain reaction (RT-PCR) and methylation-specific PCR (MSP). 100 For instance, DNA methyltransferase 1 (DNMT1) typically preserves normal methylation patterns during cell division. DNMT1 through DNA methylation downregulates Brain Expressed X-linked protein 1 (BEX1), a marker of stemness the same as EpCAM mentioned previously. Reduced DNMT1 levels result in increased BEX1 expression, which blocks RUNX3 which is a transcription factor that ordinarily suppresses β-catenin. Consequently, elevated CTNNB1 expression enhances the self-renewal properties of HCC-CSCs.77,101
Promoter methylation also affects other regulators of the Wnt/β-catenin pathway. For example, secreted frizzled-related proteins (SFRPs), which inhibit this pathway, are often silenced by promoter methylation in HCC, as seen in chronic hepatitis, cirrhosis, and HCC tissues, but not in normal liver cells. This suggests an early role in carcinogenesis. 102 Additionally, in HBV-HCC tissues, SFRP1 and SFRP5 are downregulated via DNMT1-mediated methylation, driven by the HBx protein, which enhances Wnt/β-catenin signaling and contributes to HCC development. 103
Similarly, Wnt inhibitory factor 1 (WIF1), which interacts with multiple Wnt ligands to downregulate the Wnt pathway, and Dickkopf-related protein 3 (DKK3), a Wnt receptor antagonist, were found by Ding et al. to be downregulated through promoter methylation in HCC. Specifically, WIF1 downregulation was observed in HCC patients who were non-cirrhotic and Hepatitis B surface antigen (HbsAg) negative. On the other hand, DKK3 downregulation was more common in older non-cirrhotic patients and those with advanced stages of HCC. These findings suggest that WIF1 and DKK3 may represent two independent mechanisms contributing to HCC development. 100
SRY-related HMG-box gene 17 (SOX17), a member of the transcription factor superfamily with a high mobility group (HMG) domain, is another negative regulator of the Wnt/β-catenin pathway. SOX17 interacts with β-catenin and TCF/LEF via their HMG domains. Jia et al. discovered that SOX17 was promoter-methylated in 82% of HCC cell lines, a modification absent in normal liver cells. This methylation led to nuclear β-catenin accumulation, and the restoration of SOX17 expression effectively suppressed carcinogenesis. 104
Finally, potassium channels, particularly potassium voltage-gated channel subfamily Q member 1 (KCNQ1), are methylated in HCC cells. Fan et al.'s gain-of-function study demonstrated that KCNQ1 overexpression in HCC reduced the cells' metastatic potential, highlighting the role of KCNQ1 methylation in promoting EMT and contributing to HCC progression. 105
Histone Modifications
Histone modifications are another critical aspect of HCC pathogenesis. The catalytic subunit of Polycomb-repressive complex 2 (PRC2), Enhancer of zeste homolog 2 (EZH2), plays a pivotal role by trimethylating histone H3 lysine 27 (H3K27me3), leading to the repression of Wnt antagonists such as AXIN2, PRICKLE1, NKD1, PPP2RB2, and SFRP5, which normally suppress Wnt/β-catenin signaling. Cheng et al. demonstrated that EZH2 activity in the promoter regions of these antagonists amplifies β-catenin signaling and promotes hepatocarcinogenesis, even in the absence of mutations in CTNNB1, AXIN1, or AXIN2.106,107
Moreover, in NAFLD- related hepatocarcinogenesis, histone deacetylases (HDACs), particularly HDAC8, have been implicated. SREBP-1, a lipogenic transcription factor, upregulates HDAC8, which interacts with EZH2 to further enhance the repression of Wnt antagonists. 108 Additionally, HDAC8 deacetylates the K63 residue of pyruvate kinase M2 (PKM2), facilitating its nuclear translocation and binding to β-catenin, thereby promoting Cyclin D1 gene transcription. 109
Non-Coding RNAs
Non-coding RNAs are functional RNA molecules that are not translated into proteins. Among these, microRNAs (miRNAs) regulate target genes by binding to the 3′-untranslated regions of messenger RNA (mRNA), while long noncoding RNAs (lncRNAs) interact with DNA, proteins, and RNA, including miRNAs, to exert their biological functions. Both lncRNAs and miRNAs are considered critical regulators in HCC, with their dysregulation contributing to tumorigenesis. 110 Specifically, the interaction between lncRNAs and miRNAs forms a competing endogenous RNA (ceRNA) network, known as the “sponge effect,” which effectively “soaks up” miRNAs, preventing them from suppressing their target mRNAs. This process leads to increased expression of genes that would otherwise be silenced by miRNAs, thereby promoting tumor growth.99,110
In HCC, this ceRNA network enhances the Wnt/β-catenin pathway by increasing the translation of target mRNAs for proteins such as Wnt3a, c-Myc, Cyclin D1, and others through various interactions between lncRNAs and miRNAs. Several specific ceRNA networks have been linked to the upregulation of the Wnt/β-catenin pathway in HCC, including the LINC00346-miR-542-3p, LINC00355-miR-217-5p, LINC00355:8-miR-6777-3p, LINC00662-miR-15a/miR-16/miR-107, LINC01278-miR-1258, and FEZF1-AS1-miR-107 networks.110-116
Additionally, exosomes, which are small vesicles secreted by cells, can contain miRNAs. In HCC, it has been shown that under hypoxic conditions, exosomes containing miR-1273f are secreted, leading to the repression of LHX6, a downregulator of the Wnt/β-catenin pathway. This exosome-mediated inhibition of LHX6 further promotes hepatocarcinogenesis. 117
In essence, DNA methylation plays a pivotal role in regulating cancer stem cell (CSC) properties and driving the carcinogenesis of HCC, presenting DNA methylation as a promising target for therapeutic intervention. Histone modifications, particularly those mediated by EZH2 and HDAC8, further disrupt Wnt/β-catenin signaling in HCC, underscoring their potential as key targets in HCC management. Additionally, non-coding RNAs, through their diverse interactions and regulatory networks, contribute to the epigenetic modulation of the Wnt/β-catenin pathway in HCC, offering several novel therapeutic avenues.
Role of Genetic Variations in β-Catenin Mutant HCC
Single nucleotide polymorphisms (SNPs) represent prevalent genetic variations within DNA sequences. Prior research has explored the potential implications of SNPs in various tumors and diseases.118,119 Here, we present a summary of studies on genetic variants affecting HCC risk and outcomes.
A study by Li et al. (2023) investigated TRIM16, known as a tumor suppressor in hepatocellular carcinoma, for genetic variants potentially influencing HCC risk and prognosis. Through gene-wide SNP analysis in TRIM16 and assessments in independent cohorts, a missense variant, rs2074890 (G > T), resulting in an amino acid change (E121D), was associated with reduced HCC risk and improved prognosis. Functional experiments revealed that TRIM16121D variant exhibited more potent inhibitory effects on HCC cell proliferation, migration, and invasion. Mechanistically, TRIM16121D enhanced binding to β-Catenin and mediated its degradation via K48-linked ubiquitination, thereby inhibiting the Wnt/β-Catenin pathway. This suggests that the TRIM16121D variant impacts HCC risk and prognosis through Wnt/β-Catenin pathway regulation, offering insights into HCC pathogenesis. 120
Ambrozkiewicz et al. (2022) aimed to investigate the impact of combined genetic mutations and immune cell densities on patient survival in HCC. CD8+ T cell density, rs2853669 polymorphism, CTNNB1 mutations, and telomerase reverse transcriptase promoter (TERTp) mutations were analyzed from tissue specimens of 67 HCC patients. TERTp mutations were prevalent but not directly linked to survival, whereas rs2853669 polymorphism and CTNNB1 mutations correlated with improved time to recurrence (TTR). High CD8+ T cell density in the tumor center and invasive margin was associated with longer TTR and disease-free survival (DFS). Combining genetic and immune factors enhanced predictive value for survival outcomes. For instance, combining CTNNB1 mutations and high CD8+ T cell density in the tumor center significantly improved TTR and DFS. 19
Chen et al. (2018) investigated the association between WNT1-inducible-signaling pathway protein 1 (WISP1) SNPs and clinicopathologic characteristics of HCC. Six WISP1 SNPs were analyzed in 332 HCC patients and 664 cancer-free controls. Their study showed that patients with specific WISP1 SNP variants had a lower risk of reaching advanced clinical stages. Additionally, a synergistic effect between specific WISP1 SNP haplotypes and alcohol consumption was observed in HCC development. These findings suggest that WISP1 SNPs may serve as markers or therapeutic targets for HCC. 121
Li et al. (2017) explored the relationship between polymorphisms in Wnt/β-Catenin signaling pathway genes (CTNNB1 and WNT2) and HCC in the Chinese Han population. Twenty tagging SNPs were analyzed in 320 HCC patients, 320 chronic HBV-infected patients without HCC (N-HCC), and 320 healthy controls. Their study showed that specific genotypes and alleles of WNT2 rs4730775 were associated with HCC susceptibility compared to healthy controls, while specific genotypes and alleles of CTNNB1 rs3864004 and rs11564475 were correlated with HCC compared to N-HCC patients. Interactions among these SNPs were significantly linked to HCC risk. Additionally, the CTNNB1 rs4135385 polymorphism was associated with higher risk for advanced-stage HCC. These findings suggest that genetic polymorphisms in WNT2 and CTNNB1 genes are closely related to HCC risk and progression in the Chinese Han population. 122
Kim et al. (2016) aimed to investigate the association between genetic variations in the Wnt/β-Catenin signaling pathway and the development, progression, and survival of HCC in patients with HBV infection. Seven SNPs in AXIN1, AXIN2, CTNNB1, and WNT2 genes were analyzed in 245 HBV-associated HCC patients and 483 chronic HBV carriers without HCC. Results showed that the CTNNB1 rs3864004 A allele was linked to a reduced risk of HCC development. Haplotype analysis revealed a higher frequency of the CTNNB1 G-A/G-A haplotype in HCC patients. Additionally, specific SNPs in AXIN1 and WNT2 genes were associated with tumor size and stage in HCC. Carriers of the AXIN1 rs214252 C allele exhibited longer survival. Multivariate analysis indicated that the absence of the CTNNB1 haplotype A-A and advanced tumor stage were independent predictors of poor survival in HCC patients. These findings suggest that genetic polymorphisms in the CTNNB1 gene may influence tumor development and survival in HBV-associated HCC. 123
Saitta et al. (2015) aimed to investigate the presence of genetic mutations in CTNNB1 and TP53 genes in hepatocellular carcinoma patients with occult hepatitis B virus infection (OBI). Tumor DNA was analyzed from 61 HCC patients, including those with OBI, HBV surface antigen (HBsAg)/OBI-negative, and HBsAg-positive cases. No mutations in CTNNB1 exon 3 or TP53 were detected in any case. However, the TP53 R72P polymorphism was found in 29.5% of cases, with no significant differences between OBI and non-OBI cases. This suggests that genetic polymorphism in TP53 can influence HCC development in HBV affected patients. 124
Studies of Genetic Variations in β-Catenin Mutant HCC.

Therapeutic Approaches targeting β-Catenin Signaling in HCC
HCC presents a distinctive challenge due to its prognosis being influenced not only by tumor staging but also by the concurrent impairment of liver function often associated with cirrhosis. Various staging systems are employed worldwide for HCC, including European systems like the French staging system, Barcelona Clinic Liver Cancer (BCLC) staging system, and Cancer of the Liver Italian Program (CLIP), as well as Asian systems such as the Okuda staging system, Japan Integrated Staging (JIS), Tokyo score, and Chinese University Prognostic Index (CUPI). Among these, the BCLC staging system stands out for its comprehensive consideration of tumor status (tumor size, number, presence of vascular invasion, and extrahepatic spread), liver function (evaluated by Child-Pugh class), and patient performance status (assessed by ECOG classification and symptomatology). Extrahepatic metastases in HCC commonly involve lymph nodes, lungs, and bones, typically assessed using PET/CT imaging or a combination of chest and abdominal CT scans with a bone scan. 125 Current treatment options for HCC encompass hepatic resection, transplantation, transarterial chemoembolization, and systemic therapies tailored to the stage of disease progression. In recent years, significant advancements have been made in managing HCC, with multiple systemic therapies now approved for the treatment of unresectable HCC. The combination of atezolizumab and bevacizumab has emerged as a promising first-line treatment, demonstrating improved progression-free survival compared to single-agent tyrosine kinase inhibitors like sorafenib and lenvatinib. Additionally, the phase III HIMALAYA trial recently revealed that the combination of durvalumab and tremelimumab also increased overall survival compared to sorafenib. With ICIs becoming more prevalent in various cancer treatment settings, comparing different immunotherapy combinations could further refine treatment approaches for specific cases. Second-line therapies, including regorafenib, cabozantinib, and ramucirumab, are also integral to HCC management.126-129 Despite these advancements, challenges persist in HCC therapy due to its complexity and the influence of factors like timing of diagnosis and comorbidities. Understanding the tumor microenvironment and its signaling pathways remains crucial for effectively targeting HCC cells with particular emphasis on the Wnt/β-Catenin signaling pathway due to its important role in HCC.130-132
Research across various cancers involving the Wnt/β-catenin pathway has led to the development of numerous molecules targeting different steps within this pathway. Some of these effects were discovered serendipitously with drugs initially used for other indications, including indomethacin, sulindac, rofecoxib, peruvoside, aspirin, celecoxib, pyrvinium, pirfenidone, propofol, and sevoflurane with the latter two having their action by downregulating miR-25-3p.55,133-136
Conversely, other drugs have been specifically designed to disrupt Wnt/β-catenin signaling. Microbial derivatives such as PKF115-854, CGP049090, PKF115-584, PKF222-815, and PKF118-310 are examples of such molecules that inhibit the binding of TCF to β-catenin in vitro 137 . In a study by Wei et al., PKF118-310, PKF115-584, and CGP049090 demonstrated high cytotoxicity against HCC cell lines both in vitro and in vivo, while exhibiting significantly lower toxicity toward normal liver cells, making them promising candidates for chemotherapy. 138
Another target within the Wnt/β-catenin pathway is the cAMP-responsive element-binding protein (CREB-CBP), which acts as a transcriptional co-activator by binding to β-catenin. It has been found that ICG-001 can disrupt this interaction, effectively hindering cancer growth in vitro in both colon and pancreatic cancers.139-141 It has been shown that IC-2 a derivative of ICG-001 and Pri-724 both have anti-tumor effect in vitro on HCC cells. 142
Gene therapy holds great promise for cancer treatment, driven by advances in our understanding of the molecular pathways involved in cancer development. Over the years, various molecules have been developed for this purpose, with small interfering RNA (siRNA) and antisense oligonucleotides (ASOs) being among the most commonly used or currently in development. These RNA-based drugs function by binding to mRNA or pre-mRNA, thereby preventing the translation of target proteins 143 In both in vitro and in vivo studies, siRNA and ASOs targeting β-catenin have been shown to reduce tumor growth in CTNNB1-mutated HCC cells showing promise for treatment in HCC patients.144-146
Therapies That Targets the Wnt/β-Catenin Pathway in current and Completed Clinical Trials.

Future Directions in β-Catenin Mutant HCC research
The study of Wnt signaling in liver diseases, including HCC, grown substantially, with the number of related manuscript publications increasing annually. This rise was particularly exponential before 2015, stabilizing at approximately 180-200 studies annually from 2016 onwards. 156 Despite numerous reviews based on expert opinion, bibliometric analysis is still needed to provide a comprehensive overview, analyze current trends and predict future hotspots. 156 The advent of artificial intelligence promises to expedite and enhance such analyses.
Researchers have employed tools like CiteSpace to run these analyses, using various functions to streamline complex co-citation networks. Important metrics such as total citations in co-citation networks, centrality, burstness, and usage, were utilized to determine the importance of each node. Landmark manuscripts, such as the work by He et al. (1998) and Satoh et al. (2000), have been identified using these metrics, highlighting key aspects of the Wnt signaling pathway in HCC.157,158 The field has been clustered into nine subtopics, with the largest focusing on “liver zonation” and “HCC”. These clusters delve into metabolic zonation, Wnt signaling dysfunction in HCC, and the exploration of potential therapeutic targets. Emerging trends and prospects, such as liver fibrosis and epithelial-mesenchymal transition, have also been highlighted through novel strategies integrating artificial intelligence-based techniques. 156
Elucidating Underlying Mechanisms
Further research on β-Catenin mutant HCC can provide deeper insights into the molecular mechanisms driving tumorigenesis. Understanding how β-Catenin mutations arise and their impact on downstream signaling pathways can help in identifying targeted therapeutic approaches. For instance, dissecting the exact molecular alterations and their effects on the Wnt/β-Catenin pathway could reveal new intervention points.55,159
Investigating Downstream Targets
Identifying downstream targets of β-Catenin signaling in HCC can discover new therapeutic targets. By mapping the genes and pathways activated by β-Catenin mutations, researchers can develop therapies that inhibit tumor growth and progression more effectively. One example was the identification of mTOR activation downstream of mutated β-catenin, which occurred due to the glutamine synthetase (GS) gene being a direct target gene of the Wnt pathway. GS increase in CTNNB1-mutated HCC led to increased glutamine levels in tumor cells that, in turn, led to mTOR activation in preclinical models and patients. Indeed, this study also showed the efficacy of mTOR inhibitors in notably decreasing tumor burden in β-catenin mutated mouse models. 97 Another possible strategy to impair β-catenin signaling in HCC would be by inhibiting β-catenin’s interaction in the nucleus with the TCF family of transcription factors or with Histone Acetylases like CBP, many of these are currently tested in clinical trials.149,160,161
Interaction with the Tumor Microenvironment
Studying the interaction between β-Catenin mutant signaling and the tumor microenvironment can provide insights into the complex dynamics of tumor-stroma interactions in HCC. Understanding how β-Catenin mutant influences the tumor microenvironment and vice versa can reveal potential therapeutic targets and combination therapies that may enhance treatment efficacy.71,162
Development of pre-clinical Models
The development and use of pre-clinical models of β-Catenin mutant HCC are essential for new therapeutic strategies. These models can replicate key disease characteristics and provide valuable platforms for testing targeted therapies.163,164
Genetically engineered mouse models (GEMMs) are a cornerstone of oncology research, offering a precise replication of human cancer by introducing specific genetic mutations in mice that mirror those found in human tumors. GEMMs are superior to other models, such as cell line-derived xenografts (CDX), which involve implanting cultured cancer cells into mice but fail to fully capture the complexity of tumor microenvironments. Unlike patient-derived xenografts (PDX), where human tumors are transplanted into immunocompromised mice, GEMMs maintain an intact immune system, allowing for the study of tumor development within a natural physiological environment. This ability to capture the complexities of tumor microenvironments, initiation, progression, and immune interactions makes GEMMs particularly valuable for evaluating novel cancer therapies and understanding drug resistance mechanisms.
Additionally, GEMMs surpass carcinogen-induced models, which rely on exposure to carcinogenic substances to induce random mutations and tumor development. While carcinogen-induced models are useful for studying environmental cancer risk factors and spontaneous tumor development, GEMMs provide more precise control over specific genetic alterations, making them a superior tool for investigating the genetic basis of cancer and conducting preclinical drug testing. This comprehensive advantage positions GEMMs as a critical resource for advancing cancer research and improving therapeutic strategies. 165
A notable GEMM study by Galarreta et al. enhanced our understanding of β-catenin mutations in HCC. It revealed that HCCs in this model evaded the immune system by upregulating the β-catenin pathway via CTNNB1 mutations, and were resistant to PD-L1 inhibitors. This model also helped clarify some the mechanisms behind this resistance, showing impaired recruitment of dendritic cells due to decreased CCL5 expression. Tao et al. created another GEMM using the Sleeping Beauty transposon system, demonstrating that co-expression of h-MET and CTNNB1 drove HCC development. Similarly, a study by Qiao et al., using a CRISPR/Cas9-based GEMM, showed that the loss of Axin1 along with c-MET mutations activated the β-catenin pathway, leading to HCC. These advancements in GEMM technology offer valuable insights into HCC biology and therapeutic approaches, advancing our understanding and treatment of the disease.166-169
Conducting clinical Trials
Translating preclinical findings into clinical practice through clinical trials is crucial. Evaluating the efficacy and safety of targeted therapies in patients with β-Catenin mutant tumors can lead to improved treatment outcomes for this subset of HCC patients. Tailoring therapies based on individual tumors’ molecular characteristics can enhance treatments’ precision and effectiveness.170,171
For instance, many molecules targeting the Wnt/β-catenin pathway have proven to be either unsafe or ineffective in clinical trials. However, some treatments, such as monoclonal antibodies against DKK1, have shown promise in certain cancers. This underscores the need for continued research and clinical investigation to enhance patient care in oncology. 147
Overall, further research on β-Catenin mutant HCC promises to advance our understanding of the disease and develop more effective targeted therapeutic strategies. By continuing to elucidate the molecular mechanisms, investigating downstream targets, studying tumor microenvironment interactions, developing robust clinical models, and conducting rigorous clinical trials, the field can significantly improve outcomes for HCC patients.
Conclusion
HCC presents a significant global health challenge, with its multifaceted etiology encompassing viral infections, metabolic conditions, and environmental factors complicating diagnosis and treatment. The Wnt/β-Catenin signaling pathway stands out as a pivotal element in the pathogenesis of HCC, playing a critical role in tumorigenesis and influencing the tumor microenvironment. Mutations in the β-Catenin coding genes are prevalent in HCC and are associated with specific clinical characteristics, impacting both carcinogenesis and patient outcomes.
Advancements in Imaging techniques and genetic studies have enhanced our understanding of β-Catenin mutations in HCC, providing valuable insights into their role in diagnosis and prognosis. Targeting the Wnt/β-Catenin pathway offers promising therapeutic potential. However, the challenge remains in developing targeted treatments due to the pathway’s crucial role in normal physiological processes.
Future research endeavors are directed toward unraveling the molecular mechanisms underlying β-Catenin mutations, identifying downstream targets, and conducting rigorous clinical trials. These efforts aim to translate preclinical findings into clinical applications, paving the way for improved treatment options. Ultimately, the goal is to advance personalized medicine approaches in HCC management, offering new hope for more effective and tailored patient therapies.
Footnotes
Acknowledgment
Figure 1 has been created by the author and incorporates images from Servier Medical Art, which is licensed under a Creative Commons Attribution 4.0 Unported License by Servier (![]() ).
).
Author Contributions
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Anwaar Saeed reports consulting or advisory board role with AstraZeneca, Bristol-Myers Squibb, Merck, Exelixis, Pfizer, Xilio therapeutics, Taiho, Amgen, Autem therapeutics, KAHR medical, Autem therapeutics and Daiichi Sankyo; institutional research funding from AstraZeneca, Bristol-Myers Squibb, Merck, Clovis, Exelixis, Actuate therapeutics, Incyte Corporation, Daiichi Sankyo, Five prime therapeutics, Amgen, Innovent biologics, Dragonfly therapeutics, Oxford Biotherapeutics, Arcus therapeutics, and KAHR medical; and participation as a data safety monitoring board chair for Arcus therapeutics. Satdarshan Monga is on scientific advisory board or is a consultant for Vicero Inc., Mermaid Bio, UbiquiTx, and Alnylam Pharmaceuticals. He also has or has had sponsored research grants over last year from Alnylam and Fog Pharmaceuticals.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.