
Abstract
Superficial rhabdomyosarcomas (RMS) represent a rare and diagnostically challenging group of malignant neoplasms that may arise in, involve, or metastasize to the skin. Although RMS is the most common soft tissue sarcoma of childhood, cutaneous and superficial presentations are uncommon and encompass a heterogeneous spectrum of clinicopathologic scenarios. These include primary cutaneous RMS of conventional subtypes, newly recognized molecularly defined entities such as TFCP2-rearranged RMS, rhabdomyosarcomatous transdifferentiation of primary cutaneous malignancies, and metastatic RMS to the skin. Each of these entities may closely mimic a variety of benign and malignant cutaneous tumors, leading to potential diagnostic pitfalls. This review provides a comprehensive overview of superficial RMS, with emphasis on histopathologic features, immunophenotypic profiles, molecular alterations, and key differential diagnoses relevant to dermatopathologists and surgical pathologists. Conventional primary cutaneous RMS subtypes—including alveolar, embryonal, pleomorphic, and spindle cell/sclerosing variants—are discussed alongside emerging entities such as FUS/EWSR1::TFCP2-rearranged RMS, which frequently demonstrate epithelial marker expression and ALK overexpression. In addition, rhabdomyosarcomatous transdifferentiation of melanoma and squamous cell carcinoma is reviewed, highlighting the importance of clinicopathologic correlation and molecular testing to avoid misclassification. Cutaneous metastases of RMS, though exceedingly rare, are also addressed. Recognition of these diverse presentations requires integration of morphologic assessment, immunohistochemistry, and increasingly, molecular diagnostics. Continued clinicopathologic studies and molecular characterization will be essential to refine classification, improve diagnostic accuracy and enhance understanding of the biologic behavior of superficial RMS.
Keywords
Introduction
Rhabdomyosarcoma (RMS) is a malignant mesenchymal neoplasm characterized by a skeletal muscle phenotype. It represents the most common soft tissue sarcoma in individuals under the age of 20 and thereby accounts for up to 3% of pediatric cancers.1–3 Its prognosis is highly variable depending on many factors including patient demographics, tumor site, likelihood of successful resection, and extent of disease.2,4 While RMS most often arises in the head and neck (∼40%), other common sites include the genitourinary system, extremities, orbit, thorax, and trunk.5–8 Currently, there are 4 main RMS subtypes recognized by the fifth edition (2020) of the World Health Organization including embryonal RMS, alveolar RMS, spindle cell/sclerosing RMS, and pleomorphic RMS; however, the classification of RMS is rapidly evolving with additional subtypes undergoing active investigations. 9
Superficial RMSs are rare, and the most common variants include cutaneous involvement of one of the recognized RMS subtypes (listed above), primary cutaneous RMS, transdifferentiation of a primary cutaneous tumor to RMS, metastatic RMS to the skin, and only very rare other scenarios. This article aims to provide a comprehensive review of superficial RMSs, focusing on diagnostic considerations relevant to pathologists, and to encourage further research into their clinical and molecular characterization.
Primary Cutaneous RMS of Conventional Subtypes
Primary cutaneous RMS is rare with only a few case reports and small case series reported in the literature.3,8,10–16 Most tumors are of alveolar type with exceptionally rare lesions of embryonal and pleomorphic types.3,8,10–16 While primary cutaneous RMS are rare, involvement of skin from a deep RMS through direct extension is more common.10,12,15
Clinical Features
Primary cutaneous RMS have been reported in patients of various ages with a possible predilection for men.10,14 These tumors typically arise in sun-exposed skin of the face and scalp, however reports of tumors arising in the skin of the extremities, chest, anogenital region, and shoulder exist.10,12 Notably, involvement of the head and neck is most consistently seen in pediatric patients, while the tumors reported in the adult population have a highly variable site distribution.3,8,10,13,14 Clinically, these tumors vary in presentation and are rather indistinctive, ranging from a benign-appearing erythematous nodule to an indurated plaque.3,8,10–15 Congenital primary cutaneous RMS in neonates have been reported and are considered aggressive with only few reports of long-term survival. 13 Notably, primary cutaneous RMS may be misdiagnosed as a benign entity, both clinically or histologically, but often rapidly progress in size and/or metastasize prompting further evaluation.10,14 Treatment typically consists of resection with adjuvant chemotherapy and/or radiotherapy.10,11,15 Prognosis is poor, especially in patients where there is delayed treatment due to initial misdiagnosis, with several reports of local recurrence, aggressive metastatic disease, and death despite resection and adjuvant therapy.10,14
Alveolar Primary Cutaneous RMS
Alveolar RMS comprises approximately 20% to 30% of RMSs and typically affects adolescents and young adults.17–19 It has a poorer prognosis than embryonal RMS, with lower survival rates and a higher likelihood of metastasis at diagnosis.9,17 Alveolar primary cutaneous RMS is the most common type of primary cutaneous RMS and has been reported in a variety of age groups with primary sites including the extremities and the head and neck.3,10–15
Histopathologic and Immunohistochemical Features
Alveolar primary cutaneous RMS histologically consists of a proliferation of round-to-polygonal shaped atypical cells arising from the dermis or subcutis with abundant eosinophilic cytoplasm and eccentrically located hyperchromatic nuclei.3,10–15 Characteristically, the lesional cells are often arranged in nests or sheets with thick fibrovascular septa and with central loss of cellular cohesion creating the alveolar-like appearance (see Figure 1A-C). 14 High mitotic activity, focal necrosis, and wreath-like giant cells are often seen.3,10–15 In addition, a second population of atypical spindle cells may be observed.10,11,14 In most tumors, lesional cells demonstrate a myogenic immunophenotype with strong and diffuse immunohistochemical reactivity with desmin, MYOD1, and importantly myogenin, a notable differentiating attribute from embryonal primary cutaneous RMS (see Figure 1D and E).3,10–15 S100 expression in the epithelioid or spindle cell component has been seen in a few tumors and one reported tumor expressed neuron-specific enolase.10,13,15 These tumors are negative for smooth muscle actin, keratins, EMA, HMB-45, and melan-A.10–12,15
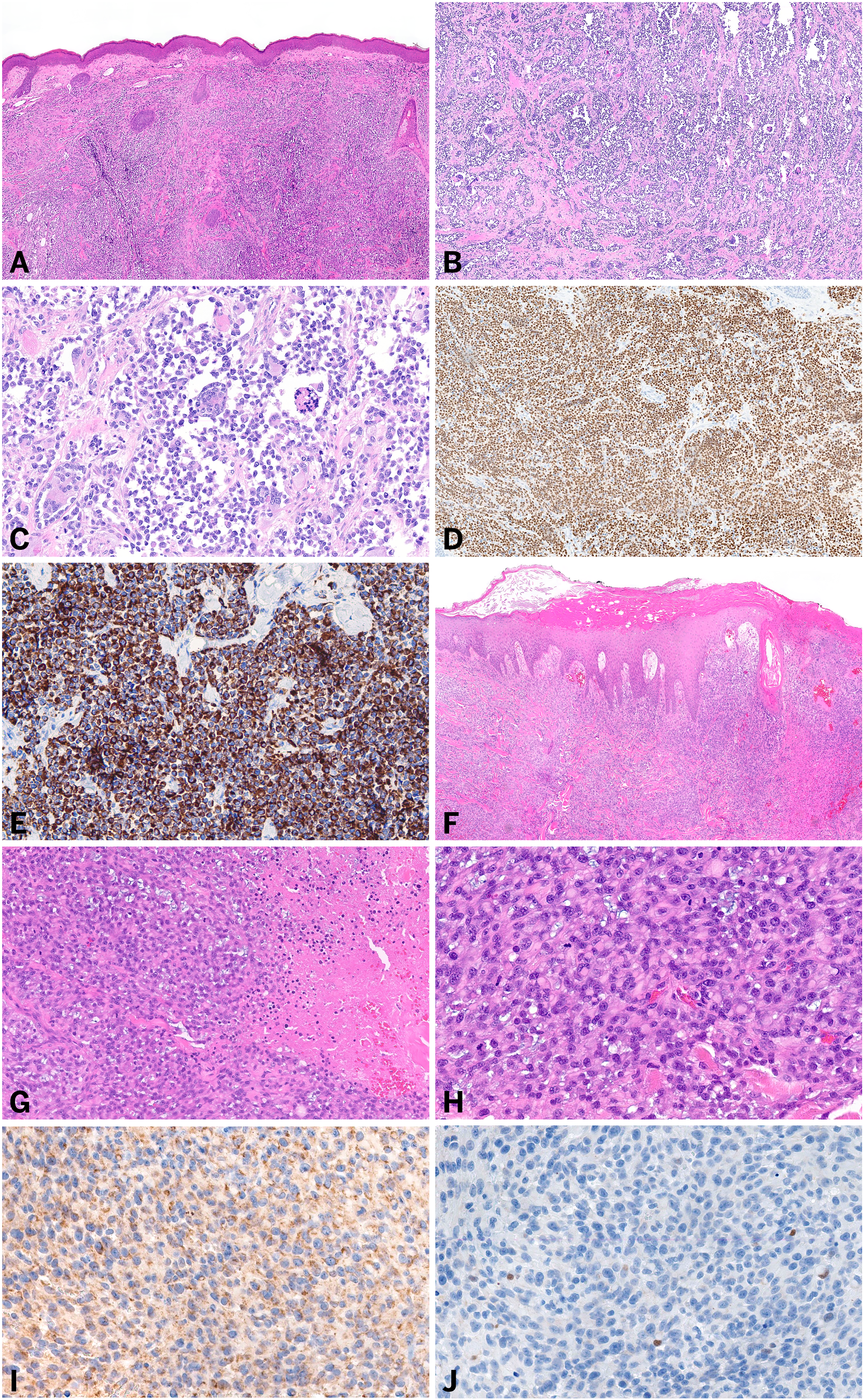
Histologic and immunohistochemical features of primary cutaneous rhabdomyosarcoma of select conventional subtypes. (A-E) Primary cutaneous alveolar rhabdomyosarcoma involving the dermis (A), composed of pleomorphic cells arranged in a characteristic alveolar-like architecture (B and C), with diffuse expression of myogenin (D) and desmin (E). (F-J) Primary cutaneous pleomorphic rhabdomyosarcoma involving the dermis (F), composed of pleomorphic oval cells with focal rhabdoid morphology and adjacent necrosis (G and H). Lesional cells are negative for MYOD1 (I, a nuclear stain) and show patchy expression of myogenin (J).
Molecular Features
Approximately 85% of alveolar RMS harbor a pathogenic PAX3::FOXO1 or, less commonly, PAX7::FOXO1 fusion.17,20 However, specimens of alveolar primary cutaneous RMS in which no characteristic fusion were identified have been reported. 14
Embryonal Primary Cutaneous RMS
Embryonal RMS is the most common RMS subtype in children typically affecting those under 5 years old with a predominance in male patients.20–22 It usually arises in the head and neck region or genitourinary tract and has a relatively favorable prognosis, with a 5-year survival rate of 70% to 85%.21–23 Embryonal RMS includes 3 main morphologic variants: classic, botryoid, and spindle cell.20,24 Embryonal primary cutaneous RMS has been predominately reported in the pediatric population and almost exclusively in the head and neck region.8,10,14
Histopathologic and Immunohistochemical Features
Embryonal primary cutaneous RMS typically consists of primitive appearing ovoid-to-spindled cells with hyperchromatic nuclei, inconspicuous nucleoli, and indistinct cytoplasm.10,14 Notably, there are often alternating areas of hypercellularity and loose myxoid stroma which leads to a distinctive impression on low power examination.8,10,14,25 Occasionally, more conspicuous rhabdomyoblastic differentiation can be observed in rare cells as evidenced by abundant eosinophilic cytoplasm and rarely, even cross striations may be visible. By immunohistochemistry, myogenic markers such as Myo-D1, myogenin, and desmin are positive but often more focal and less diffuse than alveolar primary cutaneous RMS, in particular the myogenin expression.8,10,12,14,25
Molecular Features
Molecular analysis of embryonal primary cutaneous RMS has not identified a fusion or genetic hallmark, but the lack of FOXO1-related fusions and/or the presence of mutations in RAS, TP53, or BCOR may be supportive of embryonal primary cutaneous RMS over other RMS subtypes.20,24
Pleomorphic Primary Cutaneous RMS
Pleomorphic RMS makes up approximately 10% of RMSs; however, it almost exclusively occurs in adults with a predominance in male patients.7,22,26 Pleomorphic RMS carries a similar prognosis to alveolar RMS with reports of 5-year survival of around 27% to 50%.22,27 Only a few patients with pleomorphic primary cutaneous RMS have been reported, most occurring in older adults with sites including the scalp, extremities, cheek, and periorbital region.14,16 However, these studies did not include next-generation sequencing to assess for melanoma-associated driver mutations, and therefore, the possibility that a proportion of these tumors represent transdifferentiated melanomas with rhabdomyosarcomatous differentiation cannot be excluded. In modern practice, pleomorphic primary cutaneous RMS should therefore be approached as a diagnosis of exclusion. At minimum, essential diagnostic features should be met, including histologic features of pleomorphic cells with eosinophilic cytoplasm and immunoreactivity with desmin and myogenin. 9 It would be desirable to also have supportive clinical characteristics, such as an adult patient with specific sites as described above. Nevertheless, comprehensive immunohistochemical and molecular analysis remains critical to support or refute the diagnosis.
Histopathologic and Immunohistochemical Features
Pleomorphic primary cutaneous RMS is composed of large, highly pleomorphic, spindled or polygonal and often multinucleated cells arranged in a fascicular, storiform, or varying “pattern-less” architecture.14,16,28 Rhabdoid features, including abundant eosinophilic cytoplasm, may be seen (see Figure 1F-H).14,16,28 Lesional cells often demonstrate strong and diffuse expression of desmin; however, other myogenic markers, including Myo-D1 and myogenin, can be focal and weak (see Figure 1I and J).14,28
Molecular Features
Similar to embryonal primary cutaneous RMS, molecular findings have not been reported in pleomorphic primary cutaneous RMS. However, current knowledge of pleomorphic RMS shows a lack of a genetic hallmark. 29
Spindle Cell/Sclerosing Primary Cutaneous RMS
Spindle cell/sclerosing RMS are less common than embryonal RMS and alveolar RMS, constituting up to 10% of RMSs.22,30,31 These tumors most commonly arise in the paratesticular region, head and neck, extremities, and are rarely found intraosseous.22,30,32 Characterized by spindled and/or sclerosing morphology, this type of RMS consistently harbors pathogenic MYOD1 (L122R) and/or PIK3CA mutations.30,31,33 Although spindle cell/sclerosing RMS with VGLL2::NCOA2 fusion and other related fusions characteristic of the congenital/infantile subtype, as well as TFCP2-rearranged RMS, were initially classified within this category, emerging evidence suggests that these entities are more accurately considered distinct subtypes warranting separate consideration.34–36
Primary Cutaneous RMS with FUS/EWSR1::TFCP2 Fusion
TFCP2-rearranged RMS is a recently described subtype of RMS with a strong preference for osseous sites, particularly the craniofacial bones. However, a subset of these tumors arise in soft tissue including superficial sites such as the skin.37–41 To date, 6 lesions of cutaneous origin have been reported involving the chest, back, mid-thoracic region, flank, shoulder, and scalp.37–40 While the clinical presentation varies, tumors were described as a firm erythematous nodule, indurated erythematous plaque, morphea-like lesion, multinodular mass, or an ulcerated mass.37–41 Similar to those arising in bone, primary cutaneous TFCP2-rearranged RMS tends to arise in middle aged adults with a slight predominance in male patients.37–40 Limited knowledge of radiographic features for primary cutaneous RMS with TFCP2-related fusions exists; however, the appearance of a multilocular mass with low intensity on T-1 weighted and high intensity on T-2 weighted magnetic resonance imaging has been reported in a single case report. 37 Treatment typically consists of surgical resection with adjuvant chemotherapy and/or radiotherapy.37,39 Prognosis is often poor with reports of high-grade malignant transformation, frequent local recurrence, lymph node involvement, and death of disease.37–39
Histopathologic and Immunohistochemical Features
Histologic features of this rare tumor are variable; however, the most classic pattern is that of a combination of mixed spindled and epithelioid morphology.37–40 The epithelioid tumor cells tend to be enlarged, with prominent nucleoli, nuclear pleomorphism, and frequent mitotic figures within a myxoid to collagenous stroma (see Figure 2A-D).37–40 Multinucleated giant cells and zones of necrosis may be seen.37–39 These tumors tend to show focal to diffuse expression of skeletal muscle markers, in particular MYOD1, with more limited desmin and rare myogenin expression which led to their classification of RMS.37–40 Notably, in some reports, MYOD1 was only expressed in the round epithelioid tumor cells and was negative in the spindle cell component. 37 Importantly, the lesional cells are also often strongly reactive with epithelial markers (e.g., keratin AE1/AE3 and keratin CAM5.2). Another very useful and potentially therapeutically relevant marker is ALK, as a significant subset of tumors show diffuse ALK expression (see Figure 2E-H).37–41 One possible explanation for this phenomenon is aberrant ALK activation, which may occur through several mechanisms, including the presence of ALK fusions (in addition to the TFCP2 fusion) or internal genomic deletion, but studies further investigating this finding remain limited.42,43 Some of the reports also mentioned cytoplasmic CD30 expression, predominantly in the high-grade epithelioid components.37–40 EMA, CD56, and ERG have shown weak or partial positivity in few tumors. 38
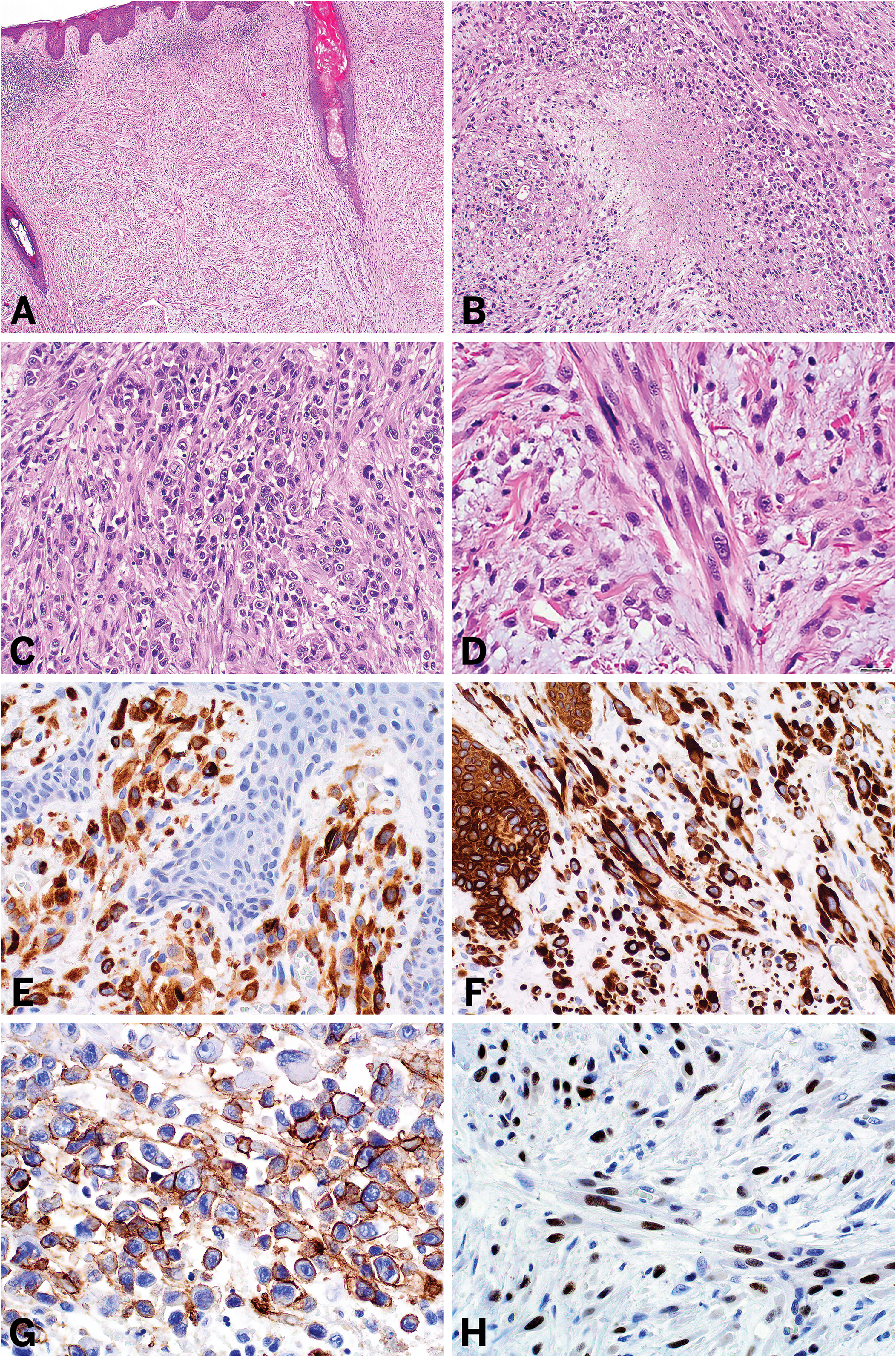
Histologic and immunohistochemical features of primary cutaneous rhabdomyosarcoma with FUS/EWSR1::TFCP2 fusion. Sarcomatoid lesion arising within the dermis (A) composed of mixed epithelioid (B and C) and spindle (D) cell morphology with focal necrosis (B) within a myxoid to collagenous stroma. Lesional cells express ALK (E), keratin AE1/AE3 (F), CD30 (G), and MYOD1 (H). *Photographs (A-H) courtesy of Dr Isidro Machado and Dr Sílvia Bagué from Spain
Molecular Features
Molecular analysis will demonstrate FUS/EWSR1::TFCP2 fusion. 38 Reports of recurrent FUS::TFCP2 gene fusions have typically involved exon 6 of FUS and exon 2 of TFCP2, with some lesions also demonstrating reciprocal TFCP2::FUS (exon 1 to exon 7) transcripts.39–42 Additionally, these tumors frequently show partial amplification of the 5′ ALK region and ALK related fusions. 42
Notable Diagnostic Considerations and Pitfalls
Due to the rarity of primary cutaneous RMS with TFCP2-related fusions, these tumors may be misdiagnosed at initial presentation. Additionally, ALK overexpression gives rise to a diagnostic pitfall, inflammatory myofibroblastic tumor. 39 In addition to ALK positivity, these tumors sometimes demonstrate CD30 reactivity, particularly within the high grade epithelioid components, which potentially mimics anaplastic large cell lymphoma.37–39 The invasive nature of this tumor accompanied by strong positivity of lesional cells with certain keratins (ie, keratin AE1/AE3 and keratin CAM5.2) may mimic dermal epithelioid sarcoma or sarcomatoid squamous cell carcinoma (SCC). 40 Additionally, co-expression of desmin may give rise to a differential of desmoplastic small round cell tumor; however, the expression of myogenic markers refute this diagnosis. 39 Malignant triton tumor (MTT) is an important differential to include as it may show rhabdomyoblastic features and can present with a myogenic immunophenotype; however, it lacks TFCP2 related fusions.44,45 A further discussion of MTT can be seen below under “Additional diagnostic considerations for cutaneous tumors with rhabdomyoblastic differentiation.” While additional diagnostic considerations exist, the key to correctly identifying these tumors is to pursue molecular profiling for fusion analysis. 38
Rhabdomyosarcomatous Transdifferentiation of Melanoma and Squamous cell carcinoma
Transdifferentiation is a process by which a population of cells undergo phenotypic transformation to that of a different lineage. 46 Many tumors, especially in the head and neck, have been reported to demonstrate rhabdomyosarcomatous transdifferentiation including malignant peripheral nerve sheath tumors (MTT), undifferentiated thyroid carcinoma, carcinosarcoma of the salivary gland, teratocarcinosarcoma, malignant teratoma, liposarcoma, and olfactory neuroblastoma. 47 Additionally, several common malignant tumors of the skin are known to have the potential to develop rhabdomyosarcomatous transdifferentiation, morphologically and immunophenotypically.46–49
Malignant Melanoma With Rhabdomyosarcomatous Transdifferentiation
Malignant melanoma with genuine rhabdomyosarcomatous differentiation is rare, with fewer than 20 patients reported; however, the true incidence may be higher with our continuously improving understanding of the transdifferentiation capacity of this tumor.46,49–55
Clinical Features
Primary cutaneous melanoma with rhabdomyosarcomatous transdifferentiation clinically presents in middle aged to older adults with a predominance in male patients (∼2-3:1).49–55 These tumors occur in areas of high sun exposure (eg, face, scalp, forearm) and often present as a large ulcerated and nodular cutaneous mass, although rhabdomyosarcomatous transdifferentiation has been identified in more “classic” flat pigmented lesions.49,51–53,55 Overall reports of prognosis varies, with some studies suggesting a similar prognosis to conventional melanoma while others demonstrating a poor prognosis with more than 50% of patients developing metastatic disease, most commonly to the lungs.48,49,53,55 Treatment often involves resection with adjuvant chemotherapy, immunotherapy, and/or radiotherapy.49,53
Histopathologic, Immunohistochemical, and Molecular Features
Microscopically, the diagnosis of melanoma with rhabdomyosarcomatous transdifferentiation can be difficult, as the transdifferentiated component often consists of sheets of epithelioid-to-rhabdoid and/or spindle cells with significant pleomorphism and atypia.49,51–53,55 Multinucleated giant cells and cytoplasmic hyaline inclusions are often reported (see Figure 3A and B).51–53,55 Increased mitotic activity and focal necrosis are common.51–53 However, a key histologic clue is the presence of adjacent or overlying conventional malignant melanoma. 49 In situations where the tumor has undergone full transdifferentiation or the conventional component has ulcerated, a thorough clinical history may be helpful. Furthermore, architectural features of malignant melanoma may persist even in the transdifferentiated component, including junctional nests, melanin pigment, and tumor-infiltrating lymphocytes. 49 In addition to a rhabdoid morphologic appearance, melanoma with true rhabdomyosarcomatous transdifferentiation express myogenic immunohistochemical markers.49,53 While a mixture of melanocytic and myogenic immunohistochemical expression in tumor cells is often reported, 53 occasionally tumors may demonstrate only weak or even absent expression of melanocytic markers, including HMB-45, SOX10, S100 protein, and Melan-A (see Figure 3C-F).49,50,53,54 In those scenarios molecular testing to assess for tumor mutational burden, melanoma-associated driver mutations such as BRAF, NRAS or NF1, UV signature analysis, and/or methylation testing can be performed.56–59 Notably, PRAME may be helpful in differentiating transdifferentiated melanoma from a primary cutaneous RMS; however, additional studies are required as the data are limited.49,60 Reported tumors indicate that pleomorphic RMS and embryonal RMS may show diffuse PRAME expression, whereas alveolar RMS have generally been PRAME negative. 60 Given that most reported lesions of primary cutaneous RMS to date are of the alveolar subtype, this may be useful as a screening tool. However, PRAME expression should not be used in isolation; confirmatory molecular testing is necessary to establish the diagnosis in this context. Future studies evaluating PRAME expression in embryonal RMS and pleomorphic RMS proven to lack melanoma-associated driver mutations, thereby excluding transdifferentiated melanomas, are warranted.
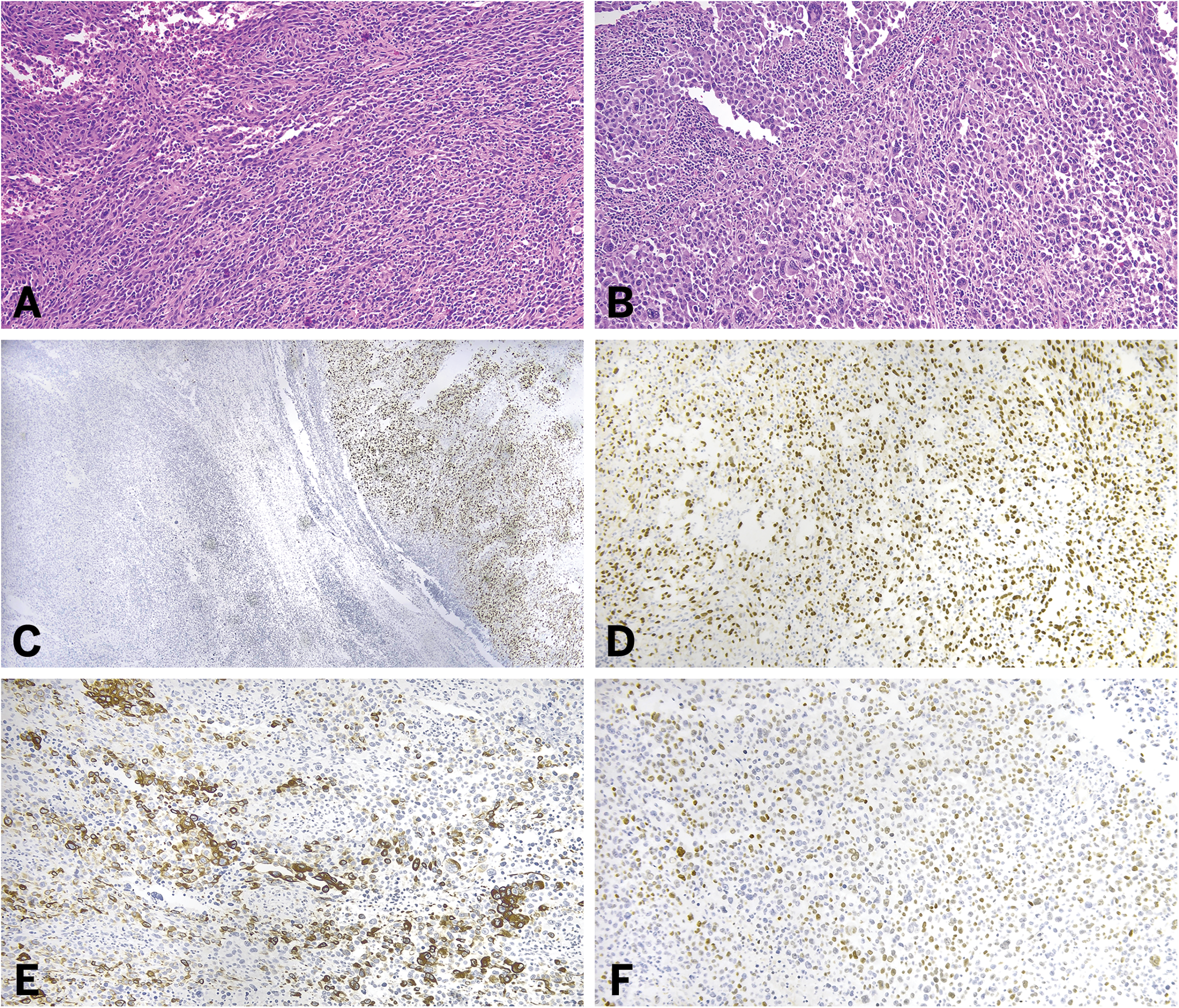
Histologic and immunohistochemical features of malignant melanoma with rhabdomyosarcomatous transdifferentiation. Sheets of pleomorphic cells with epithelioid and rhabdoid morphology (A and B) including multinucleated cells (B). Lesional cells may retain expression of melanoma markers such as SOX10 (C and D) however areas of partial to complete loss of expression can be observed (C). Characteristically, the lesional cells express myogenic markers such as desmin (E) and MYOD1 (F).
Cutaneous SCC With Rhabdomyosarcomatous Transdifferentiation
Rhabdomyosarcomatous transdifferentiation in SCC is rare with most tumors arising in the head and neck.47,61–63 Squamous cell carcinoma with spindle cell features, often called sarcomatoid carcinoma, are known to demonstrate morphologic and immunophenotypic transdifferentiation of a variety of lineages including cartilage, bone, and skeletal muscle. 47 Notably, some reports of cutaneous SCC with morphologic rhabdoid differentiation exist.64–66 However, given their lack of myogenic marker expression, these seem to represent morphologic mimics rather than representing a tumor of true skeletal muscle phenotype.
Clinical Features
Squamous cell carcinoma of the head and neck with rhabdomyosarcomatous differentiation is rare, associated with tobacco and alcohol use, and typically occurs in older adults.47,61,62 These tumors often present as a large, exophytic, ulcerated, polypoid mass arising from a mucosal site. 62 Little is currently known regarding prognostic associations with this observation.
Histopathologic and Immunohistochemical Features
On microscopic examination, rhabdomyosarcomatous transdifferentiation in SCC manifests as a population of pleomorphic polygonal or spindle cells with eosinophilic cytoplasm and an enlarged, eccentrically located hyperchromatic nucleus (see Figure 4A and B).47,61–63 Additionally, scattered bizarre giant cells may be seen and the presence of longitudinal and transverse striations have been reported.62,63 Focal necrosis may be seen. 62 As with melanomas, a key diagnostic clue in recognizing SCC with rhabdomyosarcomatous transdifferentiation is identifying an overlying SCC in-situ component or well-differentiated SCC adjacent to the sarcomatoid component.47,61–63 However, the in-situ or squamous component may be lost secondary to erosion or obliterated by the sarcomatoid tumor. 62 Immunohistochemistry can be utilized to highlight the rhabdomyosarcomatous features of these tumors as they often demonstrate strong and diffuse expression of vimentin and myogenic markers.47,61–63 Importantly, the rhabdomyosarcomatous component may retain focal or weak expression of p40, p63, and keratin (see Figure 4C-E). 62
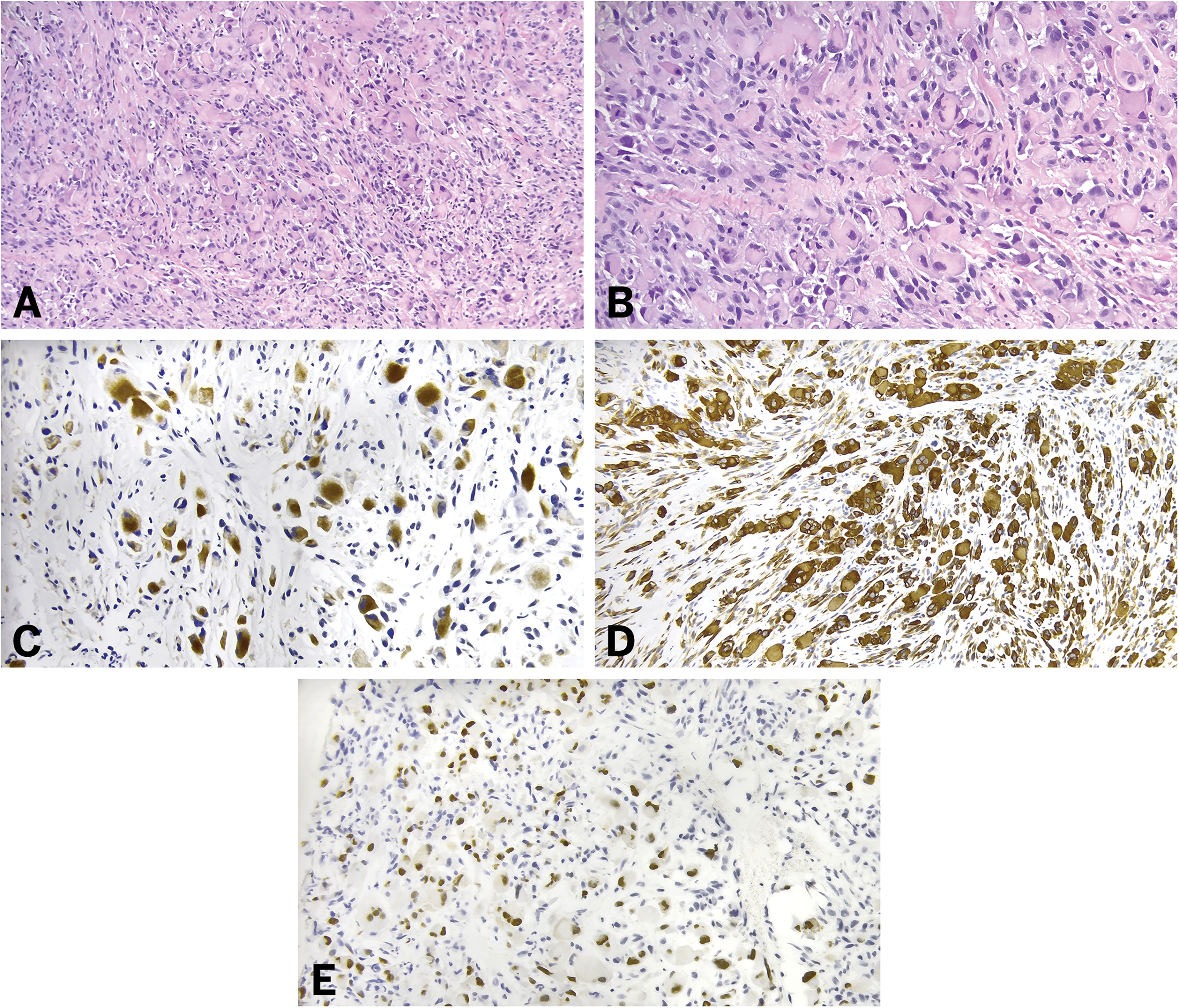
Histologic and immunohistochemical features of squamous cell carcinoma with rhabdomyosarcomatous transdifferentiation. Sheets of pleomorphic cells with rhabdoid morphology (A and B). Lesional cells show expression of epithelial markers such as keratins (C), along with aberrant expression of myogenic markers including desmin (D) and MYOD1 (E).
Metastatic RMS to a Cutaneous Site
RMSs most commonly metastasize to the regional lymph nodes and lung. 67 Alveolar RMS has additional reported metastatic sites including the brain, liver, bone, pancreas, breast, kidney, and heart.5,68,69 Spindle cell/sclerosing RMS shares similar metastatic sites. 70 However, embryonal RMS has also been reported to metastasize to soft tissues and bone marrow. Pleomorphic RMS has a very high propensity to metastasize to the lungs. 71 Rhabdomyosarcomas metastasizing to a cutaneous site is extremely rare and is more common in pediatric patients compared to adults. 72 Alveolar RMS is the most common subtype of RMS to metastasize to skin.73–76
Clinical Features
The clinical presentation of cutaneous metastasis of RMS varies and includes painless pruritic skin lesions, a single nodular fleshy cutaneous mass, “blueberry muffin” rash, and single or multiple tender firm erythematous nodules.72–78 Cutaneous metastasis most often occurs in pediatric patients. 72 Time to presentation of skin metastasis after initial diagnosis of RMS varies and has not been definitely studied, however based on existing case reports it ranges from 3 months to several years.72–78 Prognosis is poor with a 5-year survival of 20% to 30% reported in patients with metastatic RMS. 79
Histopathologic and Immunohistochemical Features
The histologic features of metastatic RMS to a cutaneous site include infiltrative nests and sheets of pleomorphic small round blue cells often involving both the dermis and subcutis.72–78 High mitotic activity is often seen.72,74–78 The tumor often exhibits the architectural pattern associated with the primary RMS subtype, such as embryonal (eg, myxoid stroma) and alveolar (eg, nested growth with fibrous septa) (see Figure 5A-D).72–78 Immunohistochemical studies aid in confirming the diagnosis, with expression of myogenic markers including desmin, muscle-specific actin reactivity, myogenin, and MYOD1.72–78 Keratin and leukocyte common antigen negativity may aid in excluding more common metastatic neoplasms of the skin (see Figure 5E-H). Notably, N-MYC amplification, with up to a 100-fold increase in copy number, has been reported in a neonate with metastatic alveolar RMS to the skin. 10
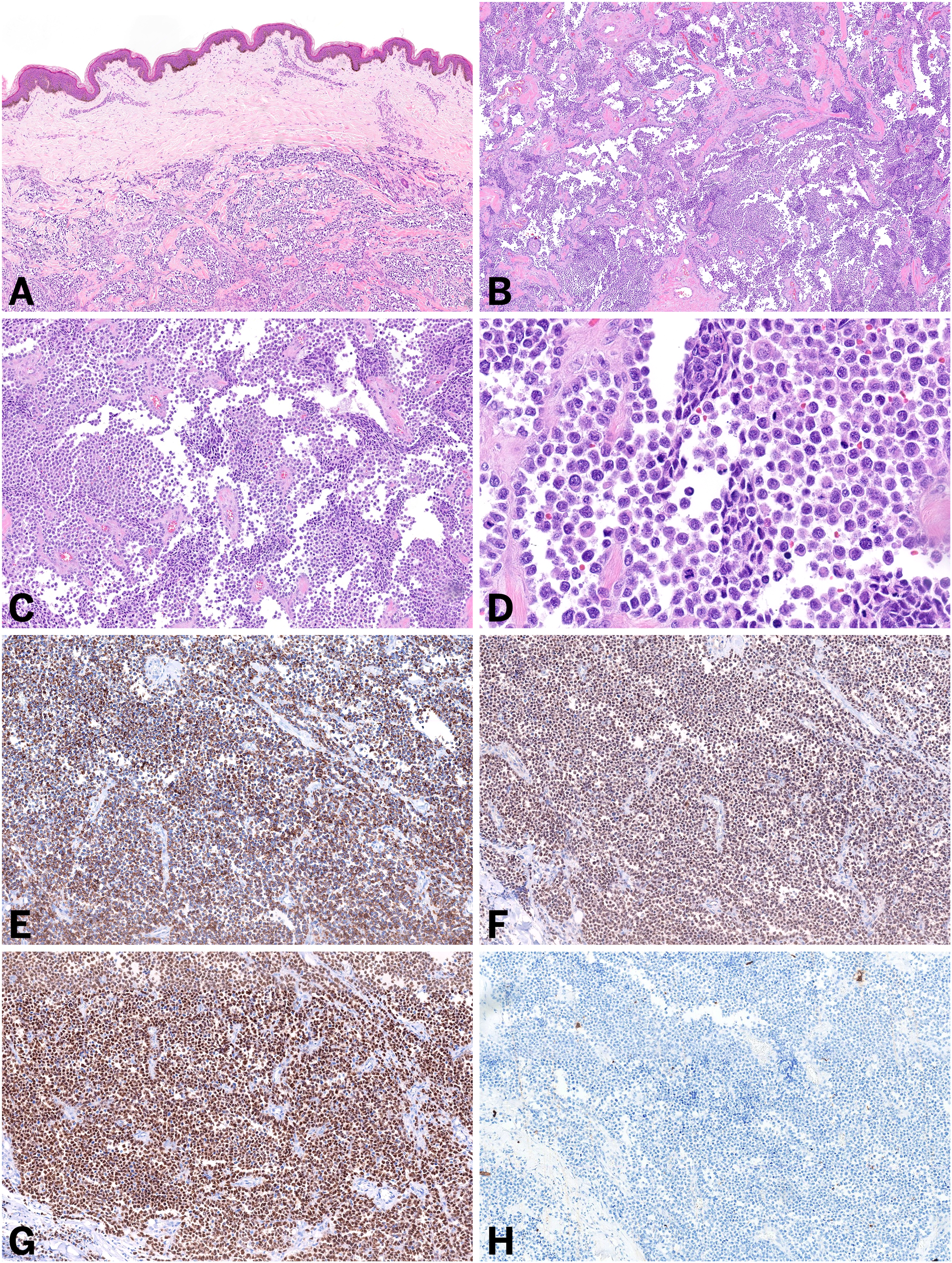
Histologic and immunohistochemical features of metastatic alveolar rhabdomyosarcoma to a cutaneous site. Infiltrative sheets of pleomorphic blue cells involving the dermis (A) with characteristic fibrous septa and central loss of cellular cohesion creating the alveolar-like pattern (B and C). Lesional cells exhibit focal rhabdoid morphology, including eccentrically located nuclei and abundant eosinophilic cytoplasm (D), and express myogenic markers including desmin (E), MYOD1 (F), and myogenin (G), while lacking keratin expression (H).
Additional Diagnostic Considerations for Cutaneous Tumors with Rhabdomyoblastic Differentiation
There is a broad differential for a cutaneous tumor with rhabdomyoblastic differentiation, including many of the entities already discussed in this review. Malignant triton tumor (MTT), which is a rare subtype of malignant peripheral nerve sheath tumor with rhabdomyoblastic differentiation, is a key or potentially challenging differential to include especially in the context of primary cutaneous RMS, cutaneous metastasis of RMS, or prototypical cutaneous neoplasms with rhabdomyoblastic differentiation (eg, SCC and melanoma).10,13,15,80 MTT often demonstrate a biphasic histologic appearance, including a spindle cell component and a rhabdomyoblastic component.44,80 Additionally, MTT often express myogenic markers, making it a challenging differential to exclude.44,80 However, MTTs often additionally show focal SOX10 expression, a marker not described in primary cutaneous RMS.45,80 MTTs typically arise in deep soft tissues, may be associated with neurofibromatosis type I, and often lose H3K27me3, which, according to current literature, is retained in primary cutaneous RMS.44,81 Additionally, molecular testing can be helpful as MTTs usually show loss of NF1, CDKN2A/B deletions, and mutations involving EED or SUZ12. 11 An additional entity which shows focal but consistent rhabdomyoblastic differentiation is the recently characterized sarcoma harboring a EWSR1::PATZ1 fusion. 82 These tumors often express myogenic, in addition to neural and epithelial immunohistochemical markers.83,84 Notably, PATZ1-related sarcomas have been reported in the central nervous system, lungs, chest wall, axilla, and head and neck; with no definitive reports of tumors arising as a primary cutaneous lesion. 82 However, the immunophenotype is a potential mimic of RMS and should be brought into the differential especially in patients where the mass may be arising from deep soft tissues and involving the skin.82–84
Conclusion
Superficial RMS, including primary cutaneous RMS, rhabdomyosarcomatous transdifferentiation of a “traditional” cutaneous neoplasm, and metastases, represent a rare and diagnostically challenging group of neoplasms that may mimic a range of primary and metastatic tumors. Recognition of histologic patterns, immunophenotypic profiles, and molecular alterations is essential for accurate diagnosis and appropriate classification. Further investigation and case reporting will be critical to elucidate the clinicopathologic spectrum of these tumors.
Footnotes
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.