
Abstract
Primary adrenal leiomyosarcoma and primary adrenal epithelioid angiosarcoma are exceptionally rare mesenchymal tumors of the adrenal gland. Both typically present as unilateral, nonfunctional adrenal masses and may closely resemble other adrenal or metastatic tumors, thus making diagnosis challenging. Immunohistochemical analysis is essential for accurate classification and clinical decision-making. We report two primary adrenal leiomyosarcomas and one primary adrenal epithelioid angiosarcoma, all occurring in elderly female patients without initial evidence of extra-adrenal disease. The leiomyosarcomas demonstrated spindle cell morphology with strong expression of smooth muscle markers and variable proliferative activity, including one patient that progressed to hepatic metastasis. The epithelioid adrenal angiosarcoma exhibited epithelioid features, extensive necrosis, diffuse CD31 and ERG positivity, and focal keratin expression. Complete surgical resection was achieved in all patients. These patients highlight the diagnostic complexity and prognostic variability of adrenal sarcomas. Documenting such rare tumors remains critical to improving diagnostic precision and guiding optimal management strategies.
Background
Primary adrenal leiomyosarcoma and primary adrenal epithelioid angiosarcoma are exceedingly uncommon malignant mesenchymal neoplasms of the adrenal gland, each constituting a rare but aggressive subset of nonepithelial adrenal tumors.1–3 In contrast to adrenocortical carcinoma and pheochromocytoma, both adrenal leiomyosarcoma and angiosarcoma are hormonally inactive and are most often discovered incidentally during imaging performed for unrelated complaints or in the workup of nonspecific abdominal or flank pain.2–4 Owing to their rarity, nonfunctionality, and overlapping radiologic features with more prevalent adrenal lesions such as adenomas, timely diagnosis remains challenging.4,5
From a histopathological standpoint, primary adrenal leiomyosarcomas, similar to their counterparts in any organ system, are characterized by intersecting fascicles of spindle cells exhibiting smooth muscle differentiation, with varying degrees of atypia and mitotic activity.2,3 Immunohistochemical (IHC) analysis is essential for definitive diagnosis, with positive staining for markers such as desmin, caldesmon, and smooth muscle actin, and negative expression of epithelial, neuroendocrine, or adrenocortical markers.4,5 In contrast, again similar to angiosarcomas arising in any organ system, primary adrenal angiosarcoma demonstrates epithelioid cytomorphology and may exhibit variable immunoreactivity for endothelial markers including CD31, ERG, and FLI1, often accompanied by focal or aberrant expression of pankeratin, further complicating differential diagnosis.6–8 Both tumors may show extensive hemorrhage and necrosis on gross examination, and proliferation indices such as Ki-67, or grading systems like the Fédération Nationale des Centres de Lutte Contre le Cancer (FNCLCC), may offer prognostic value.2,3,6
The diagnosis of epithelioid angiosarcoma in the adrenal gland is particularly challenging due to its morphologic overlap with adrenocortical carcinoma, metastatic carcinoma, sarcomatoid melanoma, and other vascular tumors including epithelioid hemangioendothelioma. As a result, a comprehensive IHC workup and meticulous clinicoradiologic correlation are indispensable to exclude metastatic disease and confirm adrenal origin.7–9 To date, fewer than 60 leiomyosarcomas and angiosarcomas arising from the adrenal gland have been reported in the English-language literature, largely as isolated case reports or small institutional series.1–3,6–8
In this study, we present two adrenal leiomyosarcomas and one adrenal epithelioid angiosarcoma, each demonstrating unique clinicopathologic features. Through detailed clinicopathological correlation and comparison with the existing literature, we aim to broaden the current understanding of these rare entities and highlight the diagnostic complexities and therapeutic considerations associated with mesenchymal tumors of the adrenal gland.
Case Report
Patient 1
A woman with a known history of hypertension and type 2 diabetes mellitus presented with right-sided abdominal pain. Cross-sectional imaging revealed a large, heterogeneous solid mass measuring 78 mm in the right adrenal region, with close anatomical proximity to the superior pole of the right kidney, duodenum, liver, and inferior vena cava (Figure 1). The lesion exhibited features suspicious for malignancy, and the patient was referred to our center for surgical evaluation.
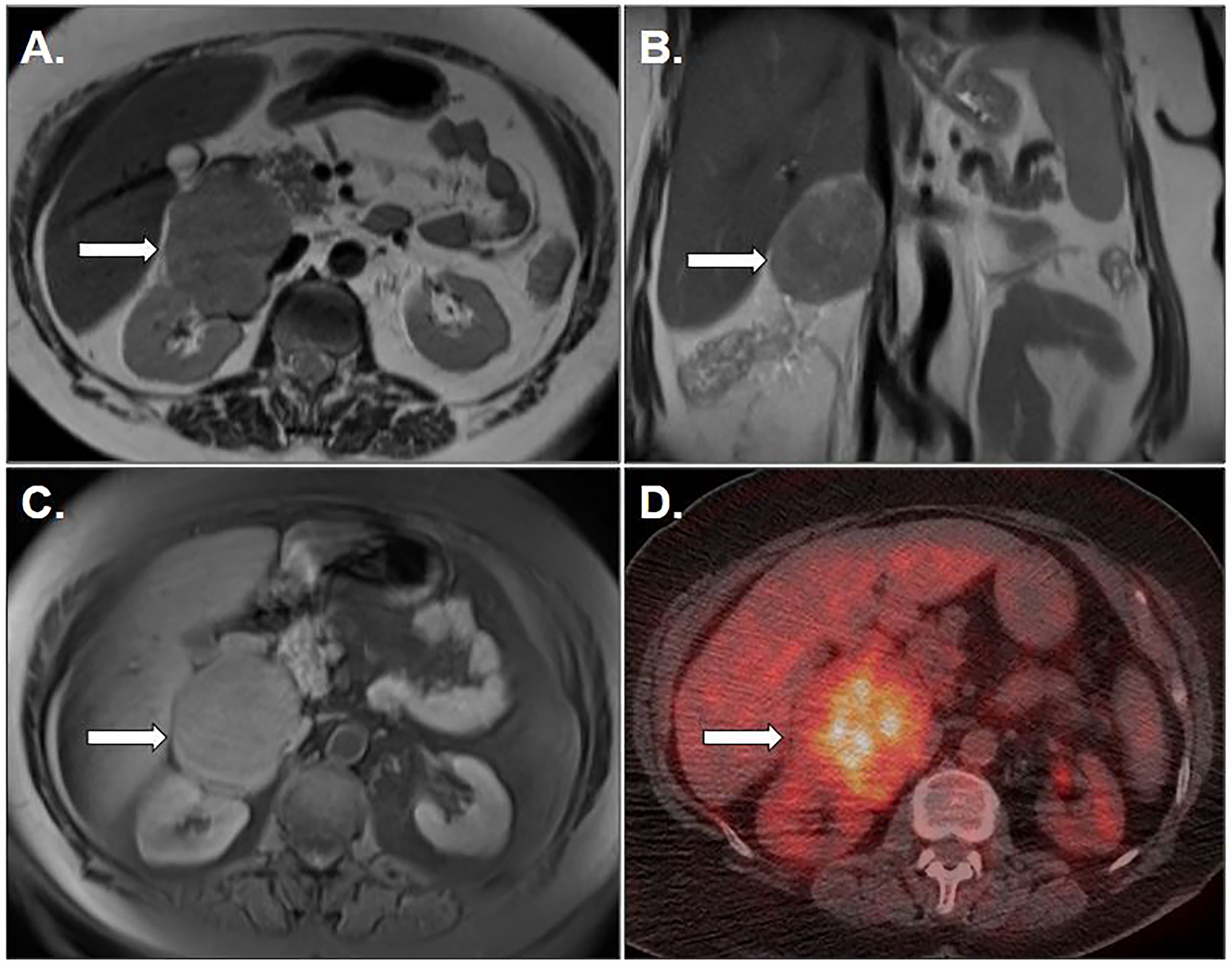
(A) Axial T2-weighted image of the upper abdomen shows a solid lesion in the right surrenal gland (arrow). (B) Coronal T2-weighted image of the upper abdomen reveals a solid lesion in the right surrenal gland (arrow). (C) Axial fat-suppressed T1-weighted postcontrast magnetic resonance image shows an enhancing right adrenal mass (arrow). (D) Fused FDG positron emission tomography/computed tomography image shows increased FDG uptake of right adrenal lesion (arrow).
Surgical resection of the right adrenal gland was performed; however, negative margins could not be achieved due to extensive adherence of the tumor to surrounding structures. Grossly, the adrenal gland was replaced by a solid, tan-white mass measuring 90 mm in diameter, which was localized within the adrenal parenchyma and exhibited focal infiltration into adjacent soft tissue.
Microscopically, the tumor was composed of interlacing fascicles of spindle-shaped cells with mild-to-moderate nuclear pleomorphism. The cellularity was high, with alternating edematous and less cellular areas. No necrosis was observed. The mitotic activity was 12 per 10 high-power fields. According to the FNCLCC grading system, the tumor received a score of 4 (differentiation: 2, mitotic figures: 2, necrosis: 0), corresponding to grade 2.
Immunohistochemistry (IHC) showed that the tumor cells were diffusely positive for caldesmon and strongly positive in focal areas for desmin and actin. The Ki67 proliferation index was approximately 30%. No pleomorphic features or multinucleated giant cells were identified, consistent with the conventional histologic subtype of leiomyosarcoma.
Due to the presence of residual tumor risk and inability to achieve negative surgical margins, the patient underwent postoperative intensity-modulated radiotherapy. Adjuvant chemotherapy was administered, consisting of 4 cycles of doxorubicin and ifosfamide. At 14-month follow-up, the patient remained alive and was under active oncologic surveillance.
Patient 2
A postmenopausal woman with a medical history of hypertension, type 2 diabetes mellitus, hyperlipidemia, hypothyroidism, and a prior thyroidectomy for multinodular goiter presented with a long-standing, nonfunctioning left adrenal mass. This lesion was initially discovered during evaluation for flank pain several years ago. Ultrasonographic imaging at that time revealed a 57-mm hypoechoic solid lesion in the left adrenal gland, which was interpreted as an adenoma and monitored radiologically.
The patient subsequently underwent total abdominal hysterectomy for abnormal uterine bleeding, revealing an endometrial polyp along with a single 5-mm intramural uterine leiomyoma, unrelated to the adrenal lesion. However, interval follow-up imaging revealed significant growth of the adrenal lesion more than 2 years after its initial detection. Magnetic resonance imaging (MRI) demonstrated a 75-mm mildly lobulated solid mass located in both the lateral and medial crura of the left adrenal gland. The lesion was mildly hyperintense on T1-weighted and hypointense on T2-weighted sequences, with radiologic features suggestive of malignancy. No extra-adrenal lesions were identified. Gross examination of left adrenalectomy showed a 75-mm solid intra-adrenal tumor, showing focal areas of nodular pushing-type infiltration into adjacent soft tissue. Microscopically, a high-grade spindle cell neoplasm composed of intersecting fascicles of moderately pleomorphic spindle cells with cigar-shaped nuclei and prominent nucleoli was present (Figure 2). Foci of coagulative necrosis were observed, and the mitotic figures were 22 per 10 high-power fields. Based on FNCLCC grading system, the tumor received a score of 6 (differentiation: 2, mitotic figures: 3, necrosis: 1), corresponding to grade 3.
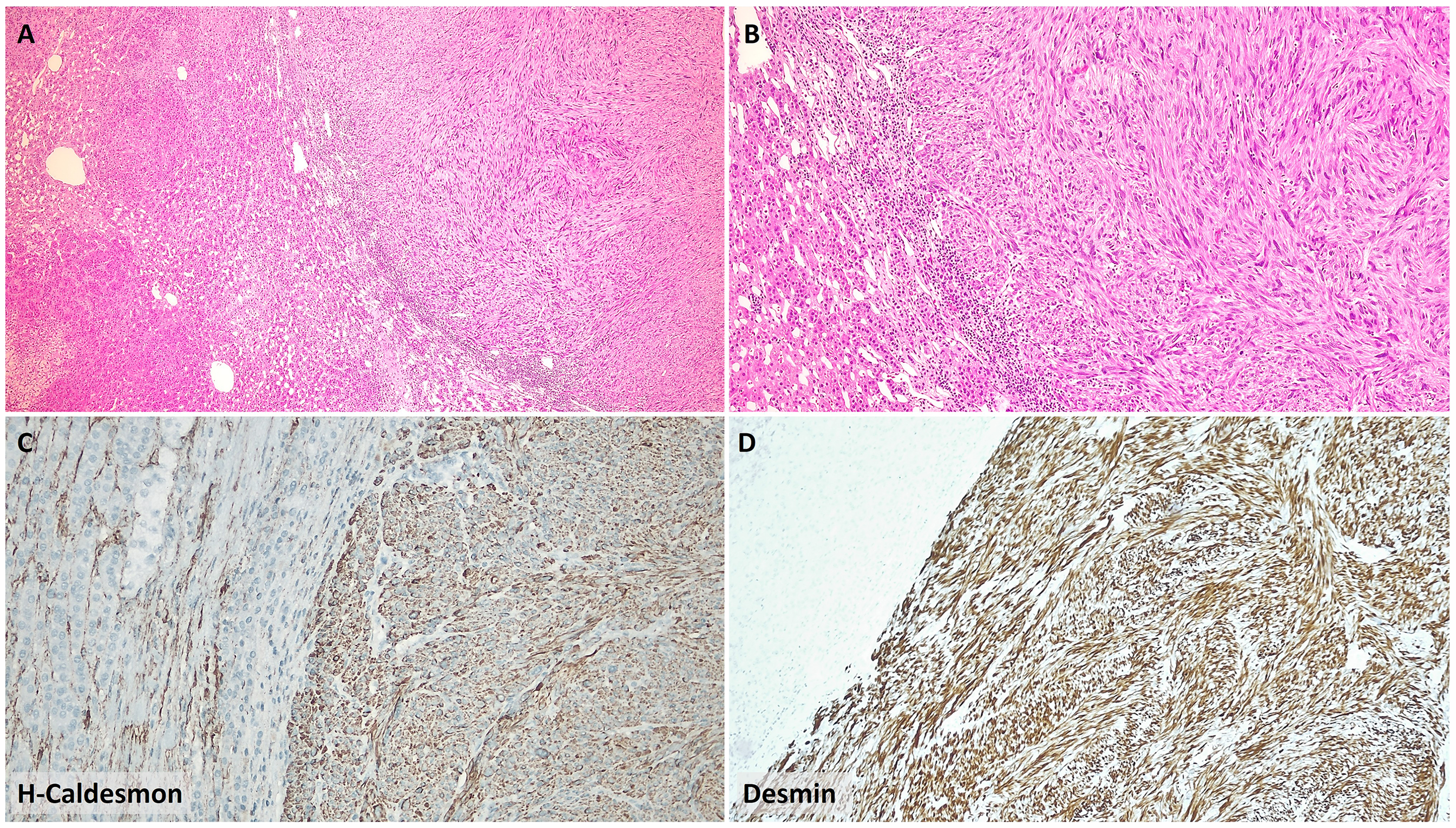
(A) A spindle cell neoplasm with expansive growth demonstrated a transition to the adjacent normal adrenal cortex (H&E, 40×). (B) Higher magnification of the same tumor showed intersecting fascicles of eosinophilic spindle cells with mild nuclear pleomorphism (H&E, 100×). (C) Tumor cells showed diffuse cytoplasmic positivity for h-caldesmon (100×). (D) Strong and diffuse cytoplasmic desmin expression was observed in tumor cells (100×).
Immunohistochemically, the tumor cells exhibited diffuse strong positivity for desmin, caldesmon, and actin. The Ki67 proliferation index was approximately 80%. No pleomorphic features were identified, and the tumor was classified as conventional leiomyosarcoma. The patient received adjuvant chemotherapy with doxorubicin and ifosfamide. During clinical follow-up for 4 years, multiple T2-hyperintense, contrast-enhancing lesions were observed in the liver, the largest measuring 22 mm, consistent with metastasis. A second-line chemotherapy regimen including gemcitabine, docetaxel, and dexamethasone was initiated. The patient remained alive and under active oncologic management.
Patient 3
An 85-year-old female patient presented to her general practitioner with nonspecific abdominal complaints and was referred to a regional hospital for further evaluation. Her medical history was unremarkable for any chronic medical or oncologic conditions. Cross-sectional abdominal computed tomography revealed a 60-mm retroperitoneal mass in the right adrenal region. No lesions were detected in other organs, and a supplementary thoracic CT scan argued against pulmonary or mediastinal involvement. Hormonal evaluation, including serum cortisol, aldosterone, and ACTH levels, was within normal limits. Urinary catecholamine metabolite levels were also unremarkable, suggesting a nonfunctional adrenal mass.
The patient underwent surgical excision of the adrenal mass. Gross pathological examination of the 70 × 70 × 50 mm specimen revealed a well-circumscribed 60 × 50 × 30 mm hemorrhagic and necrotic mass. The adrenal gland was not discernible within the specimen. Histologically, the majority of the tumor exhibited geographic necrosis. The viable areas were composed of large, anaplastic epithelioid cells with abundant eosinophilic cytoplasm, prominent nucleoli, and scattered mitotic figures (Figure 3). The architecture was predominantly solid, though focal vascular-like lumina were observed interspersed among tumor cells. Periadrenal soft tissue infiltration was present.
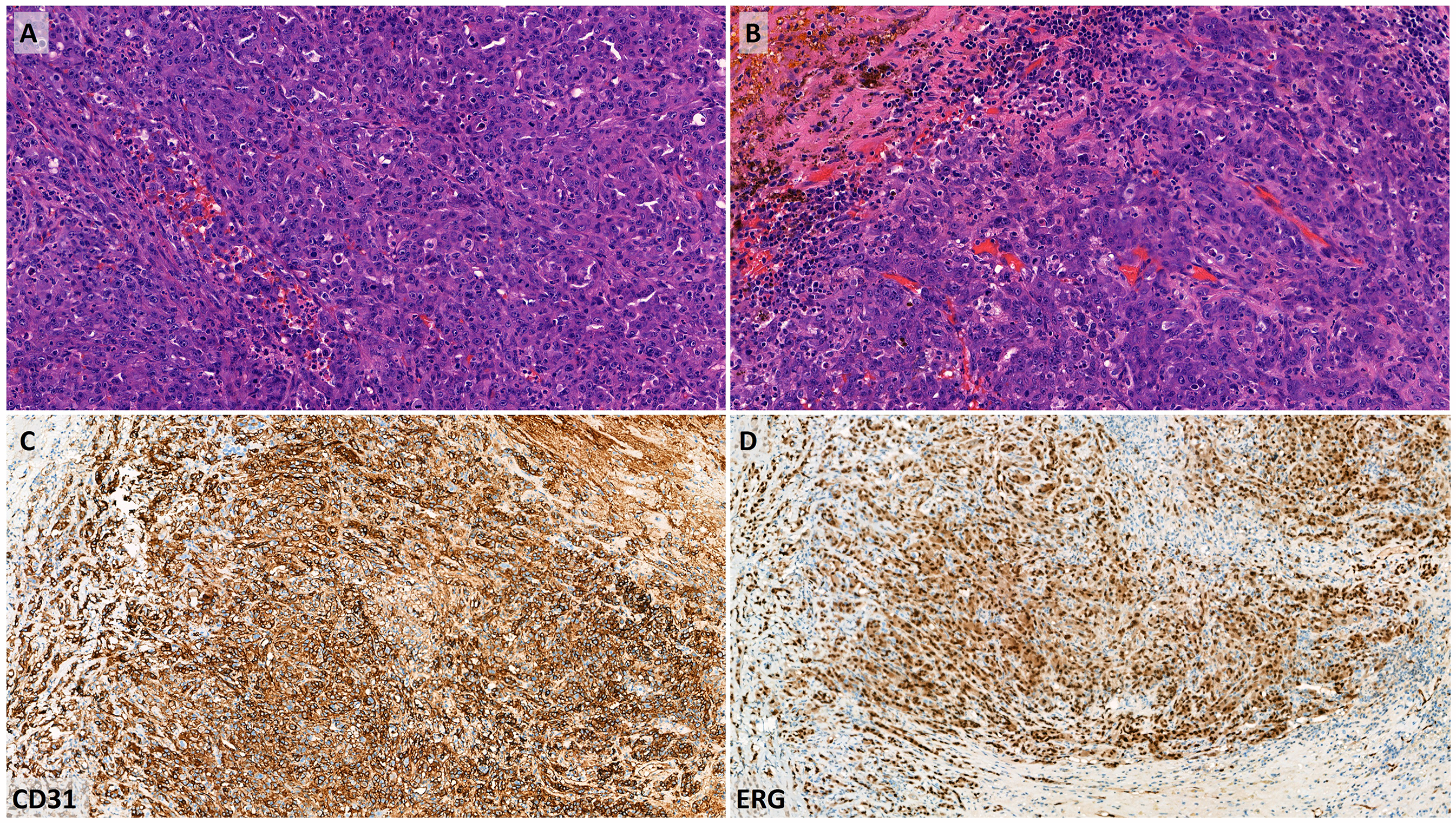
(A, B) Epitheloid high-grade malignant neoplasm with numerous mitotic figures, pleomorphism, and increased vasculature is seen in the adrenal gland (H&E, 200×). (C) Tumor cells diffusely express CD31 (100×) and (D) ERG (100×), confirming the diagnosis of epithelioid angiosarcoma.
Immunohistochemically, the tumor cells exhibited diffuse nuclear expression of ERG and strong membranous positivity for CD31, confirming endothelial lineage. Focal cytoplasmic reactivity for keratin AE1/AE3 was observed, whereas CD34 was negative. INI1 expression was retained. Notably, MYC showed diffuse nuclear positivity; however, fluorescence in situ hybridization analysis demonstrated no MYC gene amplification. Compressed adrenal cortical tissue was noted at the periphery of the mass, confirming the adrenal origin of the tumor.
Based on these histologic and immunophenotypic findings, the lesion was diagnosed as primary adrenal epithelioid angiosarcoma. The tumor was staged as pT2 according to the American Joint Committee on Cancer classification. Surgical margins were negative. Given the absence of residual disease, the patient was managed with active surveillance. Comprehensive genomic profiling is being pursued due to the exceptional rarity of the diagnosis.
Discussion
Primary adrenal leiomyosarcoma and primary adrenal epithelioid angiosarcoma share similar clinical features and anatomic localization with more common primary adrenal lesions, however, leiomyosarcoma and epithelioid angiosarcoma arising from the adrenal gland exhibit distinct histopathologic and immunophenotypic profiles. Leiomyosarcoma is composed of spindle-shaped cells arranged in fascicular patterns, consistent with smooth muscle differentiation,4–6 whereas epithelioid angiosarcoma demonstrates epithelioid morphology with vasoformative features, often accompanied by keratin expression that may mimic metastatic carcinoma.2,3,8 Leiomyosarcoma consistently shows diffuse positivity for desmin, smooth muscle actin, and caldesmon, while lacking reactivity for epithelial and adrenocortical markers.4–6 In contrast, epithelioid typically exhibits strong expression of endothelial markers such as CD31, ERG, and FLI1, with variable and often focal expression of pankeratin—a diagnostic pitfall that can lead to misclassification.2,3,8 Interestingly, MYC expression and chromosome 8 polysomy have been reported in a subset of primary epithelioid angiosarcoma patients, though their prognostic or therapeutic implications remain uncertain.2,3 In our series, both leiomyosarcomas demonstrated diffuse staining for smooth muscle markers, while the angiosarcoma displayed diffuse CD31 and ERG positivity, focal AE1/AE3 expression, and retained INI1 expression—findings consistent with previously reported epithelioid angiosarcoma profiles.2,3,8
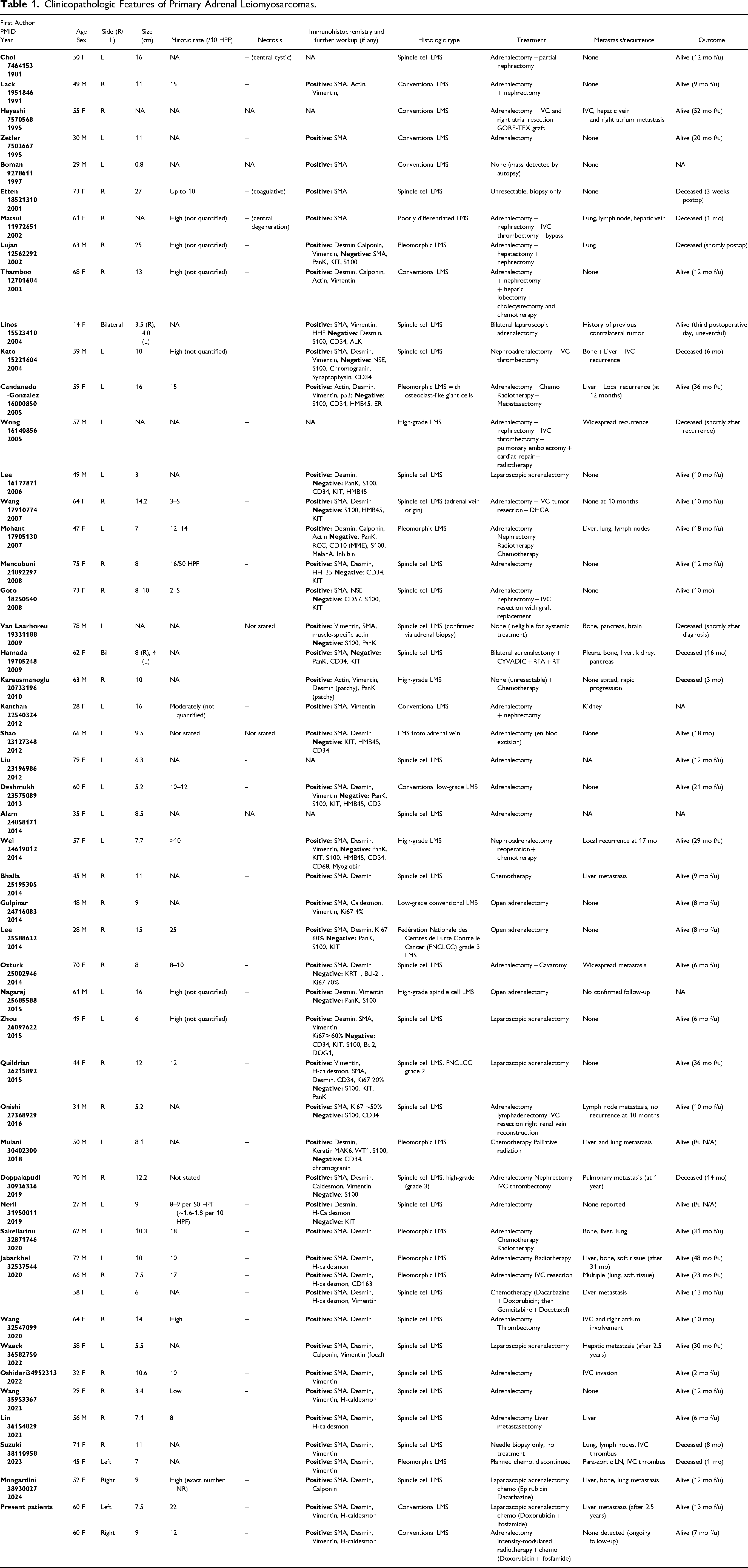
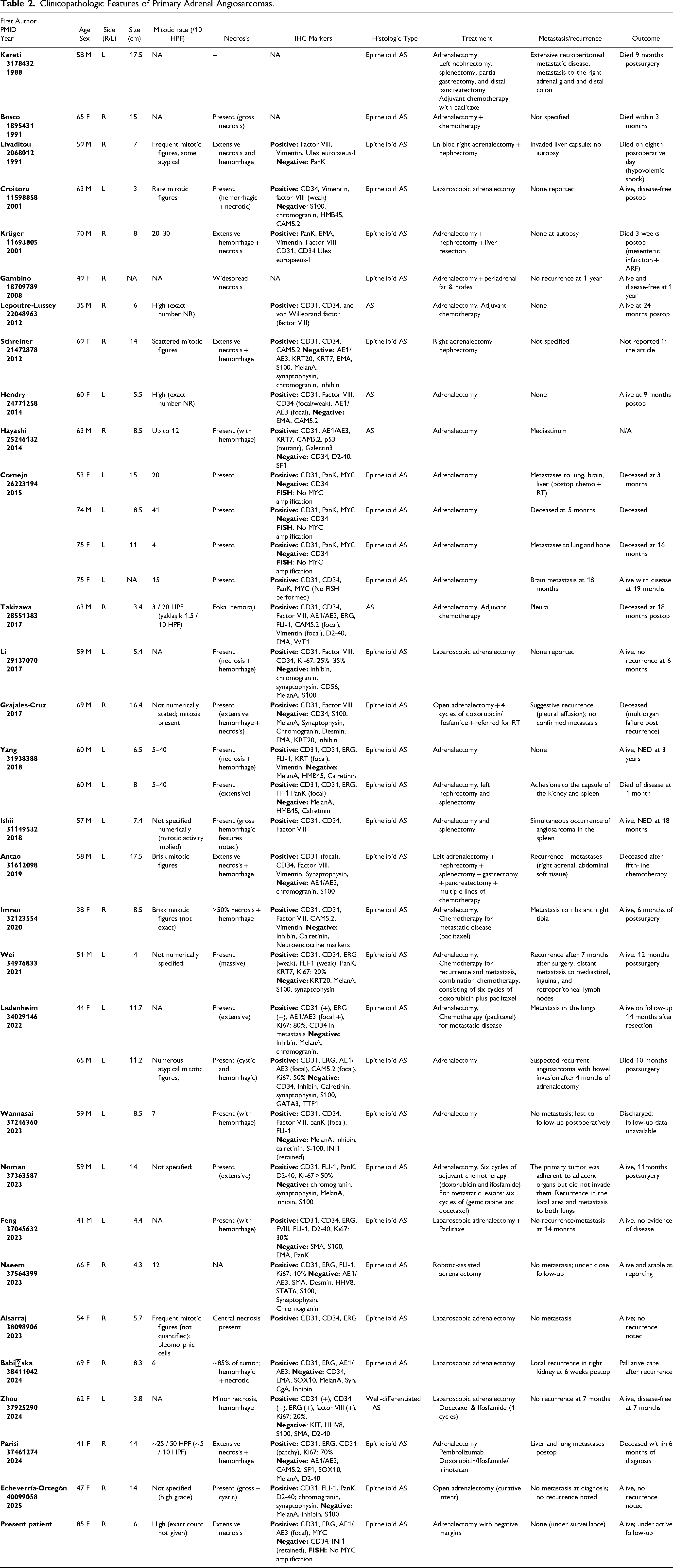
In the most extensive registry-based analysis to date, Aryal et al reviewed 332 primary adrenal sarcomas from the National Cancer Database, identifying leiomyosarcoma (37%) and angiosarcoma (27%) as the predominant histologic subtypes. 1 While informative in terms of epidemiologic patterns, the utility of this dataset is constrained by its lack of centralized histologic review and absence of granular clinicopathologic data. Critical diagnostic parameters—such as mitotic figures, necrosis, IHC profile, and margin status—were not captured. To better contextualize our findings, we conducted a comprehensive review of previously published adrenal gland leiomyosarcoma and epithelioid angiosarcoma patients, summarized in Tables 1 and 2, respectively. These tables synthesize clinicopathologic and IHC data from the largest collection of individual case reports and small series currently available in the English-language literature. Notably, the leiomyosarcoma patients demonstrated considerable variability in tumor grade, mitotic index, presence of necrosis, and Ki67 proliferation rates, all of which may influence biological behavior and prognosis. Similarly, epithelioid angiosarcoma patients exhibited wide-ranging histologic patterns, including classic vasoformative architecture as well as solid epithelioid growth, and showed diverse immunophenotypic features—particularly in regard to keratin expression and MYC status. By incorporating our three patients into this collective dataset, we aimed to provide an updated comparative resource for pathologists and clinicians managing these diagnostically complex tumors.
Clinicopathologic Features of Primary Adrenal Leiomyosarcomas.
Clinicopathologic Features of Primary Adrenal Angiosarcomas.
Prognosis remains guarded for both adrenal leiomyosarcoma and epithelioid angiosarcoma, in part due to the rarity of these tumors and the consequent lack of standardized treatment protocols.1,2,4 In leiomyosarcoma, adverse prognostic factors include high FNCLCC grade, elevated mitotic index, presence of coagulative necrosis, and incomplete surgical excision.4–6 Similarly, epithelioid angiosarcoma demonstrates a propensity for aggressive clinical behavior when associated with large tumor size, high proliferative index, extensive necrosis, and high-grade epithelioid features.2,3,8 Although complete surgical resection represents the cornerstone of management for both tumor types, the role of adjuvant therapies remains ill-defined. Limited reports have described the use of anthracycline- or taxane-based chemotherapy in epithelioid angiosarcoma, particularly in patients with recurrent or metastatic disease, albeit with variable responses.2,3,8,9 In our cohort, the grade 3 leiomyosarcoma patient developed hepatic metastases during follow-up despite adjuvant chemotherapy, whereas the patient with the grade 2 tumor remained disease-free under surveillance. The epithelioid angiosarcoma patient, whose tumor was completely resected with negative margins, is currently under active follow-up, reflecting the broader uncertainty surrounding the benefit of adjuvant therapy in such patients.
Conclusion
Primary adrenal leiomyosarcoma and primary adrenal epithelioid angiosarcoma are exceptionally rare mesenchymal malignancies that present substantial diagnostic and therapeutic challenges due to their nonspecific clinical presentation, lack of hormonal activity, and morphologic overlap with other adrenal or metastatic neoplasms. In this study, we present two additional adrenal leiomyosarcomas and one adrenal epithelioid angiosarcoma, and integrated them into an updated review of the literature to emphasize key differences and shared features in their clinicopathologic profiles, IHC characteristics, and clinical outcomes. Accurate diagnosis relies on comprehensive histopathologic and immunophenotypic evaluation, particularly in tumors with atypical or overlapping features. While complete surgical resection remains the cornerstone of treatment, the role of adjuvant therapy is not yet well defined and requires individualized decision-making. As the cumulative number of reported patients continues to grow, each new contribution offers valuable insight into the biological behavior, prognostic variables, and evolving management approaches for these rare and aggressive adrenal sarcomas.
Footnotes
Author Contributions
BYB, IC, CV, MA collected the data and BYB, MA drafted the manuscript. BYB, MA edited the manuscript, participated in the study design and coordination. All authors read and approved the final manuscript.
Availability of Data and Materials
All data generated or analyzed during this study are included in this published article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical Approval
This study was approved by the local Institutional Review Board.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.