
Abstract
Crystal-storing histiocytosis is a rare condition characterized by the accumulation of crystalline immunoglobulins within histiocytes, often associated with plasma cell disorders such as multiple myeloma. We report a 70-year-old man with a history of IgG-lambda multiple myeloma, systemic amyloidosis, and end-stage renal disease managed with peritoneal dialysis and long-term lanthanum carbonate therapy for hyperphosphatemia, who presented with gastrointestinal bleeding. Endoscopy revealed nodular erythematous mucosa in the duodenum, and biopsies showed histiocytosis in the lamina propria. The histiocytes showed abundant cytoplasm containing periodic acid–Schiff-positive diastase-resistant intracellular material. Immunohistochemical staining revealed diffuse CD68 positivity and lambda light-chain restriction of the storage material in the histiocytes, supporting a diagnosis of crystal-storing histiocytosis. Differential diagnoses, including Whipple disease, mycobacterial infections, lanthanum deposition, amyloidosis, and xanthoma, were excluded based on histological findings and clinical history. This report highlights the importance of recognizing crystal-storing histiocytosis in uncommon sites such as the duodenum, especially in the context of plasma cell dyscrasias.
Introduction
Histiocytes are immune cells derived from monocytes, playing a key role in phagocytosis and immune regulation. In normal conditions, histiocytes are found in small numbers within the lamina propria of the gastrointestinal tract, where they contribute to immune surveillance and response. 1 An abnormal increase of histiocytes, known as histiocytosis, within the duodenal mucosa can occur due to various etiologies. Infectious causes are common, with Whipple disease being a classic example, which is characterized by foamy histiocytes containing periodic acid–Schiff (PAS)-positive material due to the presence of Tropheryma whipplei. Mycobacterial infections, such as those caused by Mycobacterium avium complex, can also lead to histiocytosis, identifiable by acid-fast organisms within histiocytes. Metabolic or storage disorders may present with histiocytosis due to the accumulation of intracellular deposits. Noninfectious etiologies include plasma cell dyscrasias and autoimmune disorders. In particular, hematologic malignancies such as multiple myeloma can be associated with histiocytic proliferations containing abnormal inclusions. Crystal-storing histiocytosis is a rare condition characterized by the accumulation of crystalline immunoglobulins within histiocytes, often linked to underlying plasma cell disorders. 2
In this report, we present a 70-year-old man with a history of IgG-lambda multiple myeloma and systemic amyloidosis presenting with duodenal histiocytosis. Although histiocytosis in the duodenal mucosa often suggests Whipple disease, crystal-storing histiocytosis should also be considered, particularly in patients with plasma cell dyscrasias such as multiple myeloma.
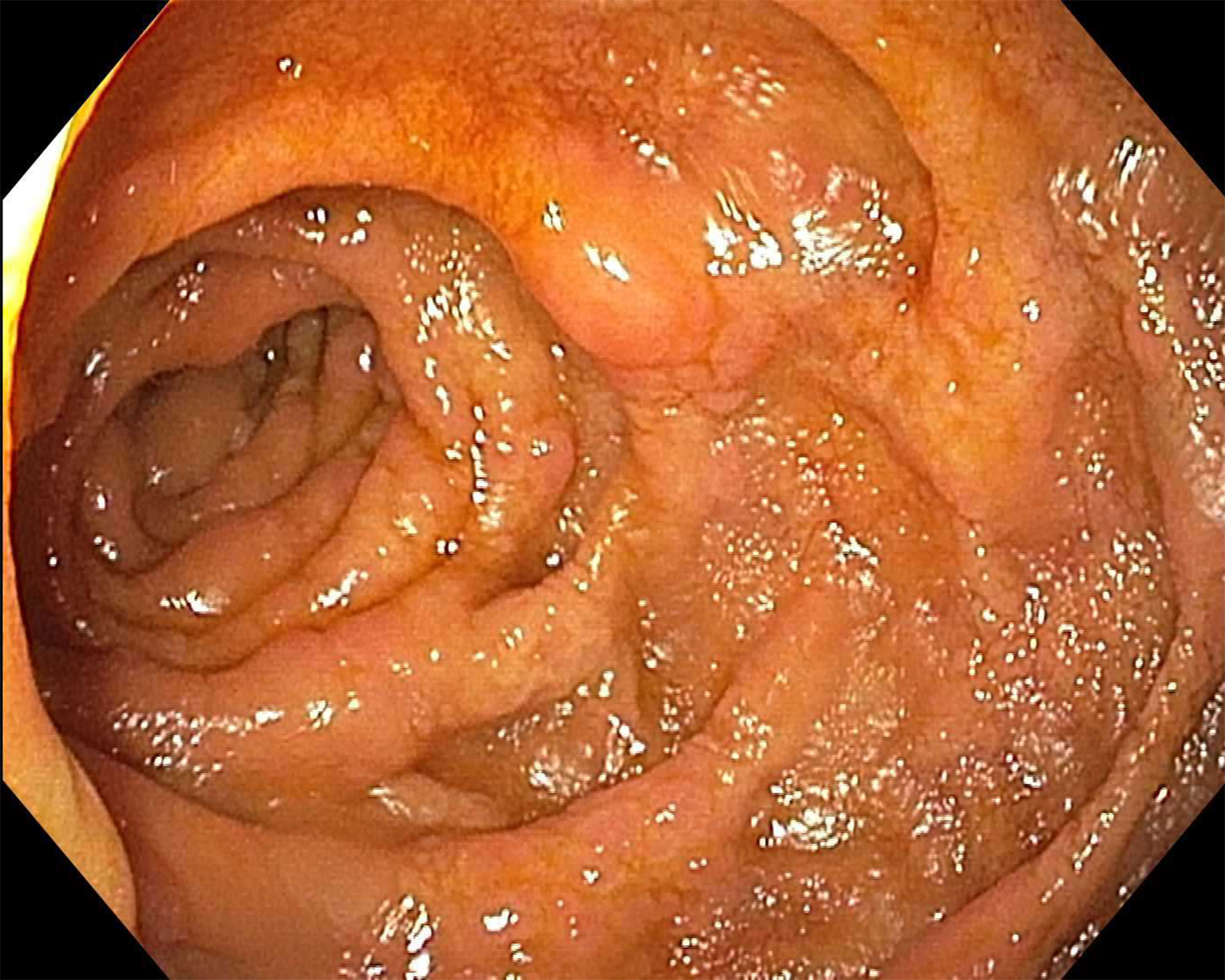
Endoscopic image of the second portion of the duodenum. The image demonstrates diffuse nodular erythematous mucosa observed in the second portion of the duodenum. Biopsies are taken from this area for histopathological evaluation.
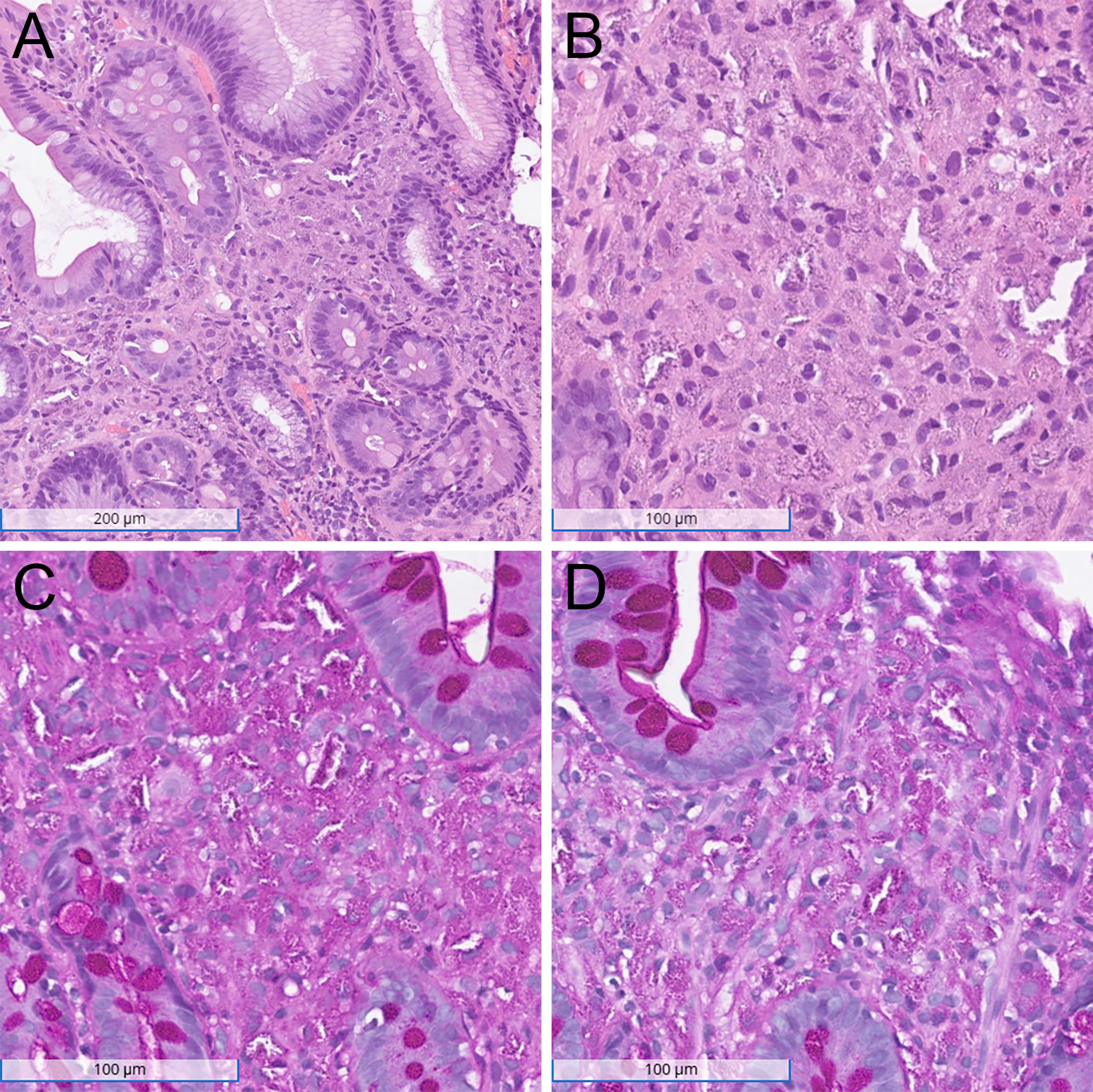
Histopathological findings of the duodenal biopsy. (A) H&E stain at low magnification (100×) shows duodenal mucosa with an expanded lamina propria filled with bland histiocytes. (B) H&E stain at intermediate magnification (200×) highlights histiocytes with abundant cytoplasm and scattered microcalcifications. (C) PAS staining demonstrates positivity within the histiocytes, indicating intracellular material storage. (D) PAS-D staining confirms the intracellular material is resistant to diastase digestion.
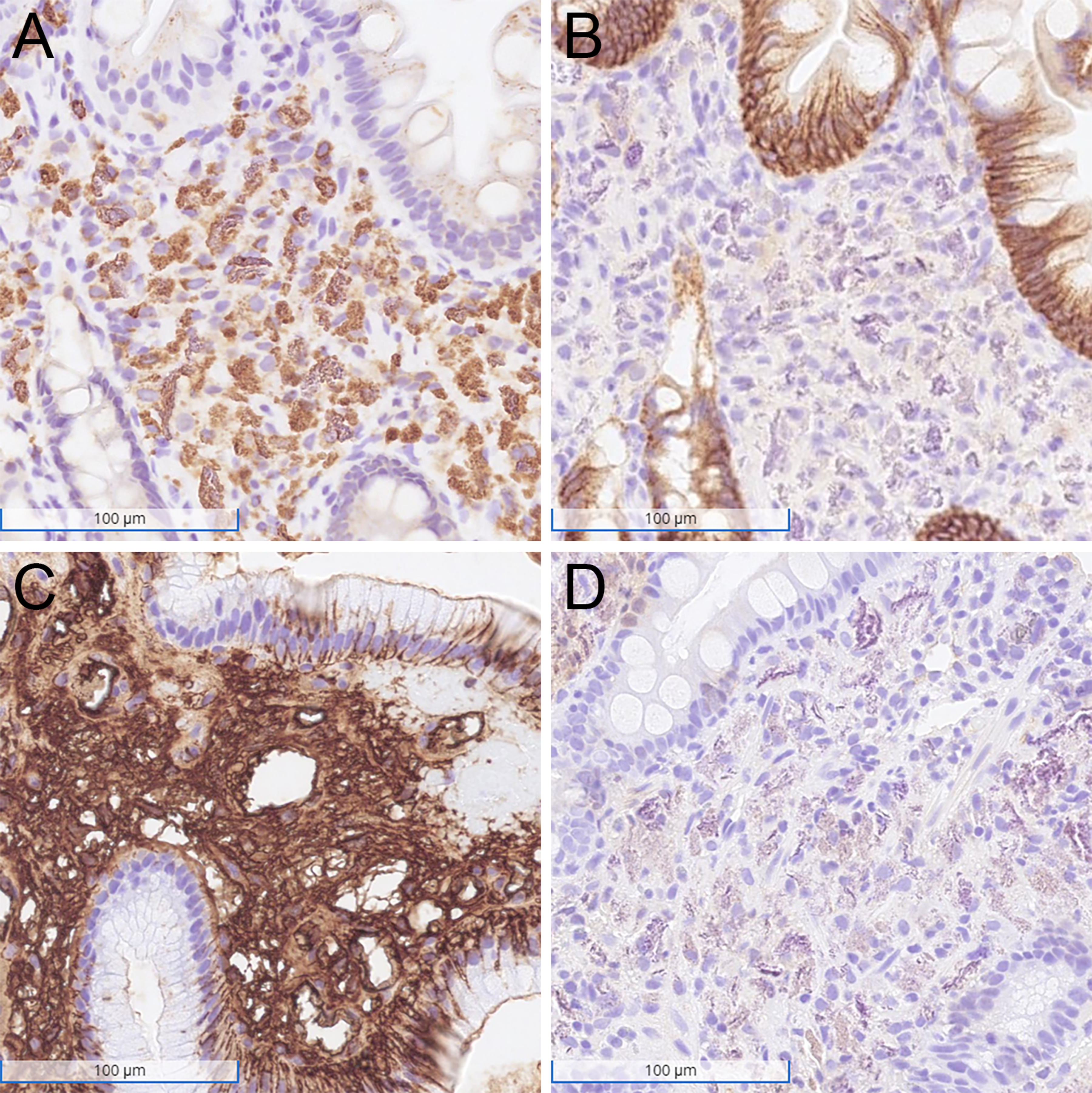
Immunohistochemical findings of the duodenal biopsy. Immunohistochemical staining for CD68 shows diffuse and strong positivity within the histiocytes (A), while CD138 is negative (B). Histiocytes demonstrate strong cytoplasmic positivity for the lambda light chain (C) and weak cytoplasmic positivity for the kappa light chain (D).
Clinical Summary
The patient was a 70-year-old man with a history of IgG-lambda multiple myeloma, systemic light chain amyloidosis, and end-stage renal disease managed with peritoneal dialysis. He had no history of autoimmune, inflammatory, or infectious gastrointestinal disorders. He had been taking lanthanum carbonate at a dose of 3000 mg/day for at least 3 years to manage the hyperphosphatemia associated with his renal disease.
The patient was initially diagnosed with multiple myeloma at the age of 66, presenting with severe anemia and acute renal failure. A bone marrow biopsy showed 36% plasma cells, and flow cytometry confirmed a monoclonal plasma cell population with lambda light chain restriction. Cytogenetic analysis revealed gains of chromosomes 1q21, 9, 13, and 15, along with a t(4;14) translocation. The serum beta-2 microglobulin level was elevated at 33.6 mg/L (reference: 0.971.84 mg/L), and the patient was staged as Revised International Staging System Stage III multiple myeloma based on this elevation and high-risk cytogenetics. 3 He underwent chemotherapy with cyclophosphamide, bortezomib, and dexamethasone, achieving a partial response with a reduction in serum M-protein by 82.3% (from 7.9 g/dL at baseline to 1.4 g/dL) after 4 cycles. A positron emission tomography-computed tomography scan performed after 4 cycles of chemotherapy revealed a 3.0 cm lytic lesion in the right lateral aspect of the C1 vertebral body, with cortical destruction and mild fluorodeoxyglucose (FDG) uptake (maximum standardized uptake value [SUVmax] = 2.9). Multiple additional lytic lesions were observed throughout the skeleton, including the spine (T1/T2, T10, and L1 vertebral bodies), bilateral pelvic bones, and the left superior acetabulum with mild FDG uptake (SUVmax = 3.8), consistent with widespread myelomatous involvement.
Over the next 4 years, the patient received multiple lines of therapy for relapsed and refractory multiple myeloma, including daratumumab-lenalidomide-dexamethasone, daratumumab monotherapy, daratumumab with cyclophosphamide and bortezomib, carfilzomib with venetoclax, and elotuzumab-pomalidomide-dexamethasone. Despite these treatments, the disease remained refractory. At age 70, the patient was admitted for a step-up dosing of Teclistamab. During this hospitalization, he developed a non-ST elevation myocardial infarction and subsequently gastrointestinal bleeding with melena and hypotension. An initial esophagogastroduodenoscopy revealed erosive gastropathy characterized by multiple localized erosions in the gastric fundus with stigmata of recent bleeding, along with congested duodenal mucosa; however, no biopsies were obtained. A repeat procedure identified a nonbleeding linear gastric ulcer with an adherent clot and diffuse nodular erythematous mucosa in the duodenal bulb and proximal duodenum (Figure 1). Clips were placed for gastric ulcer, and biopsies were obtained from the duodenum for further evaluation.
Histologic examination of the biopsy specimen revealed duodenal mucosa with patchy mild villous blunting, few foci of foveolar metaplasia, and expanded lamina propria filled with bland histiocytes containing abundant cytoplasm (Figure 2). Scattered microcalcifications were present within the lamina propria. There was no increase in intraepithelial lymphocytes or lymphoplasmacytic infiltrate in the lamina propria. Special stains for PAS revealed PAS-positive diastase-resistant intracellular material. Congo red staining confirmed amyloid deposition in the vessel walls, consistent with known systemic light chain amyloidosis. Iron staining did not demonstrate iron deposition within the histiocytes. Immunohistochemical staining showed that the histiocytes were diffusely and strongly positive for CD68, while negative for Cam 5.2, KIT, CD138, and S100 (Figure 3). Immunohistochemistry for kappa and lambda light chains demonstrated strong positivity for lambda and faint staining for kappa within the histiocytes, indicating lambda light-chain restriction. Stains for acid-fast bacilli, Fite, and Grocott's methenamine silver were negative. Additionally, a polymerase chain reaction test for Tropheryma whipplei was negative. These findings support the diagnosis of crystal-storing histiocytosis in the context of IgG-lambda multiple myeloma.
Discussion
The histopathologic findings of lambda light chain restriction within histiocytes in our patient confirm the diagnosis of crystal-storing histiocytosis, a rare disorder characterized by the accumulation of crystallized immunoglobulins within histiocytes. 2 Crystal-storing histiocytosis has been linked to various etiologies, 4 including multiple myeloma, lymphoproliferative disorders, 5 medications (eg, clofazimine and carbamazepine),6,7 autoimmune diseases including rheumatoid arthritis, 8 and infectious diseases such as Helicobacter pylori-associated gastritis. 9 However, our patient lacked clinical or pathological evidence of medication-related, autoimmune, or infectious conditions, supporting multiple myeloma as the underlying cause.
Crystal-storing histiocytosis most commonly affects the bone marrow, lymph nodes, and respiratory tracts, while gastrointestinal involvement is relatively uncommon and has been occasionally reported in the stomach.9,10 Although rare, previously reported examples of crystal-storing histiocytosis involving the duodenum have been associated with clofazimine use6,11 or underlying extranodal marginal zone B-cell lymphoma. 5 Unlike the previously reported patients, our patient had immunoglobulin-based crystal-storing histiocytosis without prior exposure to clofazimine. This highlights the importance of recognizing uncommon presentations associated with underlying plasma cell dyscrasias, considering crystal-storing histiocytosis in the differential diagnosis for the duodenum, and understanding its etiologies.
The pathomechanism underlying crystal-storing histiocytosis in multiple myeloma remains incompletely understood, but excessive production and impaired degradation of monoclonal immunoglobulin light chains play central roles. 2 Recent molecular analyses have demonstrated that pathogenic light chains frequently derive from specific germline gene segments, such as Vκ3-20 or Vκ1-39, and notably lack significant somatic hypermutation.12,13 Furthermore, detailed molecular studies have identified specific unusual amino acid substitutions, such as leucine at position 59, which may alter the protein's conformation and enhance hydrophobic interactions, facilitating crystal formation. 2 These structural alterations likely result in resistance to lysosomal degradation, leading to chronic intracellular crystal accumulation within histiocytes. Further research is necessary to clarify molecular determinants and macrophage-related factors driving crystal formation.
Although crystal-storing histiocytosis predominantly involves kappa-type immunoglobulin light chains (reported in approximately 85%), lambda-type involvement, as observed in our patient, is distinctly uncommon. 14 One potential explanation for this imbalance is the differential intracellular handling and biochemical properties of kappa versus lambda light chains. Kappa chains are more soluble within the lysosomal environment and thus may more readily crystallize and accumulate. In contrast, lambda chains typically form dimers and appear less prone to lysosomal crystallization. Our patient's rare lambda-type crystal-storing histiocytosis suggests that specific structural or biochemical properties of certain lambda chains may also predispose them to crystal formation, warranting further molecular investigation into these uncommon variants.
Differentiating crystal-storing histiocytosis from other conditions with increased histiocytes in the gastrointestinal tract requires careful evaluation. 15 Whipple disease is a primary consideration, characterized by foamy histiocytes containing PAS-positive granules. In our patient, Whipple disease was excluded by a negative PCR for Tropheryma whipplei. Similarly, negative stains for acid-fast bacilli, Fite, and Grocott methenamine silver ruled out mycobacterial and fungal infections.
Lanthanum deposition in the gastrointestinal tract is another important differential diagnosis, particularly in patients with end-stage renal disease on lanthanum carbonate therapy for hyperphosphatemia.16,17 Lanthanum deposits are often found in the stomach and duodenum and appear as dense, basophilic, granular material within histiocytes. These deposits are faintly stained with PAS and lack immunoglobulin light chain staining. 18 In contrast, the histiocytes in our patient showed moderate PAS positivity, along with strong cytoplasmic staining for lambda light chains, consistent with immunoglobulin-based crystalline inclusions. Additionally, the patient's history of IgG-lambda multiple myeloma further supports the diagnosis of crystal-storing histiocytosis over lanthanum deposition.
S-100 negativity excluded granular cell tumors and Langerhans cell histiocytosis, which are associated with S-100-positive histiocytic infiltrates. Duodenal xanthoma, another differential diagnosis, presents as foamy macrophages laden with lipid debris, often arising as a response to mucosal injury. 19 While CD68 is positive in both xanthoma and crystal-storing histiocytosis, the crystalline inclusions characteristic of crystal-storing histiocytosis and the clinical context of plasma cell dyscrasias distinguish the 2 conditions.
Amyloidosis is also an important differential diagnosis for crystal-storing histiocytosis, as both conditions can present abnormal material in the lamina propria of the gastrointestinal tract. Amyloid typically shows extracellular, vascular, and perivascular deposition but may rarely appear as round, globular eosinophilic deposits in the lamina propria, 20 mimicking crystalline inclusions. In our patient, systemic light chain amyloidosis was confirmed by Congo red staining, with classic apple-green birefringence observed under polarized light in the vessel walls. However, the histiocytic inclusions in crystal-storing histiocytosis were negative for Congo red, confirming that the crystals were not composed of amyloid. The coexistence of crystal-storing histiocytosis and systemic light chain amyloidosis in this patient highlights a potential diagnostic pitfall, as their overlapping features could lead to misinterpretation without specific staining characteristics.
The treatment and management of crystal-storing histiocytosis depend on the underlying condition, as crystal-storing histiocytosis is often secondary to hematologic malignancies. Identifying crystal-storing histiocytosis is particularly important in patients without a prior diagnosis of hematologic malignancy, as it may signal an underlying plasma cell disorder or other malignancy. Thus, crystal-storing histiocytosis can serve as an early warning—a “tip of the iceberg”—prompting further investigation for potential comorbid hematologic conditions. 21 Since our patient has already been treated for multiple myeloma, no adjustments to the treatment plan were necessary.
In conclusion, this case report highlights the importance of considering crystal-storing histiocytosis in the differential diagnosis of histiocytic infiltrates in the gastrointestinal tract, particularly in patients with multiple myeloma. Given the various underlying etiologies, including plasma cell disorders, lymphoproliferative diseases, medications, autoimmune disorders, and infections, identifying the specific cause is essential for appropriate management.
Footnotes
Author Contribution
Shunsuke Koga evaluated the clinical and pathological findings and drafted the manuscript. Zhaohai Yang provided a review and critique. Zahra Alipour conceptualized the project, evaluated the clinical and pathological findings, and provided critical review and revisions. All authors approved the final version of the manuscript.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical Approval
According to our Institutional Review Board (IRB) at the Hospital of the University of Pennsylvania, this case report does not meet the definition of human subject research requiring IRB review. This study was conducted in accordance with the Declaration of Helsinki 1975.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Informed Consent
Informed written consent was obtained from the patient.