
Abstract
Malignant peripheral nerve sheath tumors (MPNSTs) primarily originate from the neurofibromatosis 1 (NF1)- associated and/or sporadic neurofibromas. Reports of malignant transformation from a sporadic soft tissue schwannoma are rare. In most tumors documented in this context so far, the malignant component is an epithelioid MPNST with a strong and diffuse S100 positivity. We present a patient diagnosed with a sporadic soft tissue schwannoma on core biopsy, which underwent rapid malignant transformation to a spindle cell type MPNST. Somatic tumor profiling of the excision specimen revealed NF2 loss, TP53, NRAS, and subclonal RICTOR gene mutations. A retrospective immunohistochemical stain for p53 on the core biopsy demonstrated an aberrant staining pattern, indicating an early effect of TP53 in the process of malignant transformation.
Introduction
Schwann cells of neural crest origin give rise to schwannomas, neurofibromas, and malignant peripheral nerve sheath tumors (MPNSTs). Although they share a common embryonic origin, their clinical course and genetic makeup are distinct. 1 Approximately 50% of MPNSTs are associated with neurofibromatosis 1 (NF1), originating from pre-existing neurofibromas harboring germline NF1 gene mutations. Another 45% of tumors are sporadic, while the rest are radiation associated. 2 Nearly 40% of sporadic MPNSTs also show NF1 mutations and other genetic and epigenetic abnormalities that influence their biological behavior and therapeutic responses. 2
More than 90% of schwannomas are sporadic, with 77% harboring neurofibromatosis 2 (NF2) gene deletions or mutations, while some schwannomas are associated with schwannomatosis predisposition syndromes. 3 NF2 is a tumor suppressor gene that encodes Merlin (also known as schwannomin), which is highly expressed in Schwann, meningeal, and ependymal cells. Merlin inhibits the PI3K/Akt/mTOR pathway and receptor tyrosine kinase signaling, inhibiting cell proliferation, growth, and survival. Loss of NF2/Merlin leads to benign and malignant Schwann cell neoplasms. 4 It is the most common recurrent genetic alteration in sporadic and germline schwannomas. 5
Although very rare, transformation of a schwannoma to MPNST has been documented primarily in vestibular schwannomas with or without prior radiation,6–8 followed by soft tissue schwannomas (noncranial nerves).9–17 The molecular changes associated with this particular type of malignant transformation have not been well documented. We describe a patient with a biopsy-proven sporadic soft tissue schwannoma with rapid malignant transformation to a high-grade spindle cell-type MPNST with an early TP53 missense mutation, NF2 loss, and NRAS mutation, but with retained H3k27me3 and SMARCB1 protein expression.
Case Report
A 76-year-old woman who presented with low back pain was found to have a 3.5 cm complex mass within the right psoas muscle (Figure 1A and C). Biopsy of the mass showed an encapsulated spindle cell neoplasm (Figure 2A) with alternating hypercellular and myxoid hypocellular areas (Figure 2B), and hyalinized vessels (Figure 2C). The fascicles of the spindle cells showed eosinophilic cytoplasm, ill-defined cytoplasmic borders, and elongated and wavy nuclei (Figures 2D and 3A). Some cells were larger and multinucleated, and scattered hemosiderin deposition was present, consistent with degenerative atypia. Rare mitotic figures (<1/10 high-power field) were identified; however, necrosis was absent. The cellular areas were diffusely and strongly positive for S100 by immunohistochemistry (IHC) (Figure 3B), with slightly weaker staining in the myxoid areas. Most tumor cells expressed SOX10 (Figure 3C). Therefore, a diagnosis of schwannoma was made.
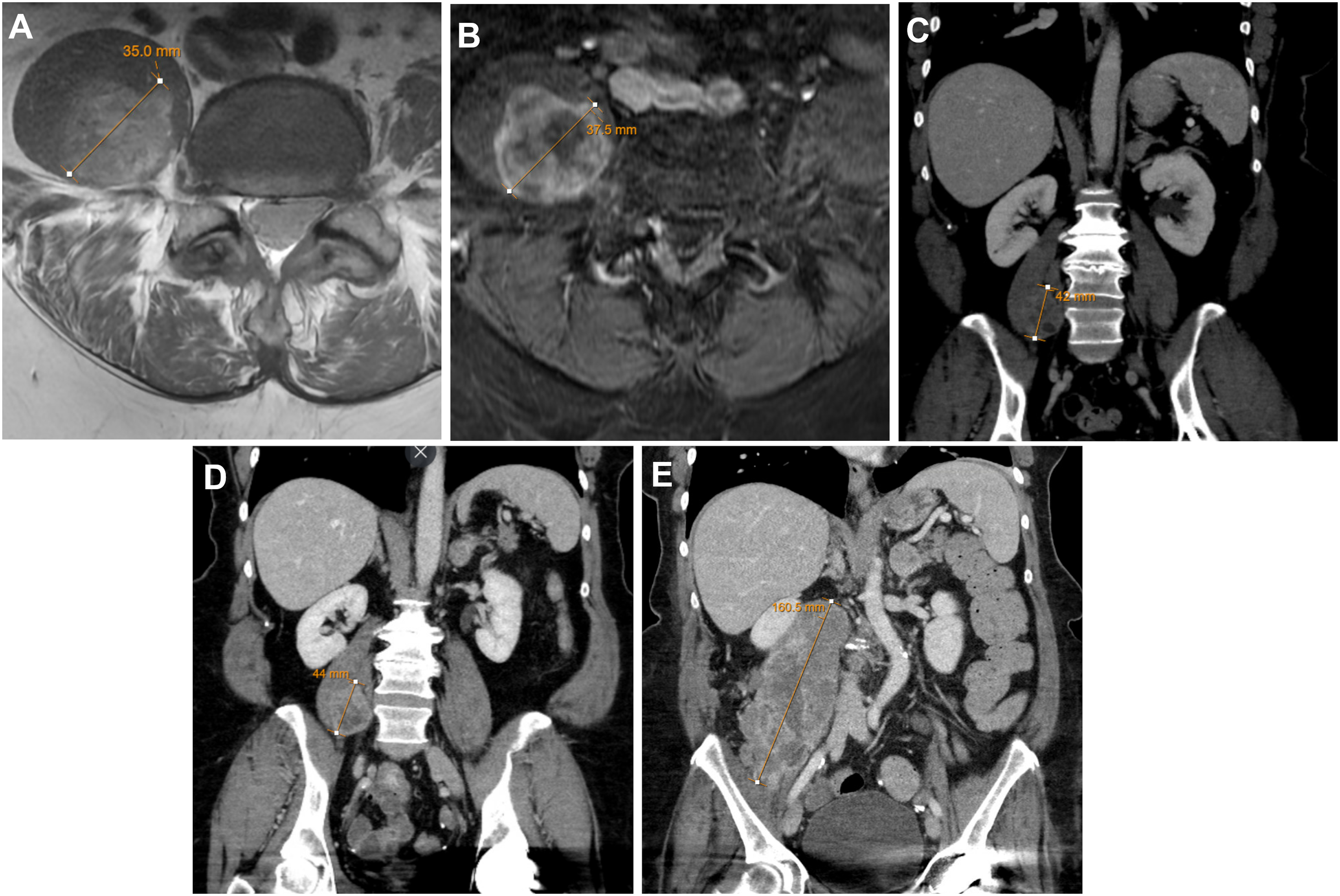
Sequential CT and MRI scans. (A) MR without contrast, at initial presentation. Within the right psoas muscle, there is a mass with heterogeneous signal intensity, predominantly hyperintense to muscle, 3.5 cm in axial dimension; (B) MR with contrast, performed 5 months later, redemonstrates the mass with heterogeneous enhancement including cystic and/or necrotic foci. It measures 3.8 cm in axial dimension, thus slightly increased in size compared with prior MRI; a change from 4.9 to 5.8 cm was observed in the craniocaudal dimension; (C) CT of the abdomen/pelvis with contrast performed 10 days after the initial MR shows an ill-defined heterogeneously enhancing lesion (4.2 cm in greatest dimension) within the right psoas muscle; (D) Postbiopsy CT reveals a slight increase in the size of the right psoas lesion; and (E) CT following attempted surgical resection of the lesion demonstrates mass-like “disfigurement” of the psoas muscle with heterogeneous attenuation/enhancement measuring 16 cm in craniocaudal dimension. Abbreviations: CT, computed tomography; MRI: magnetic resonance imaging.
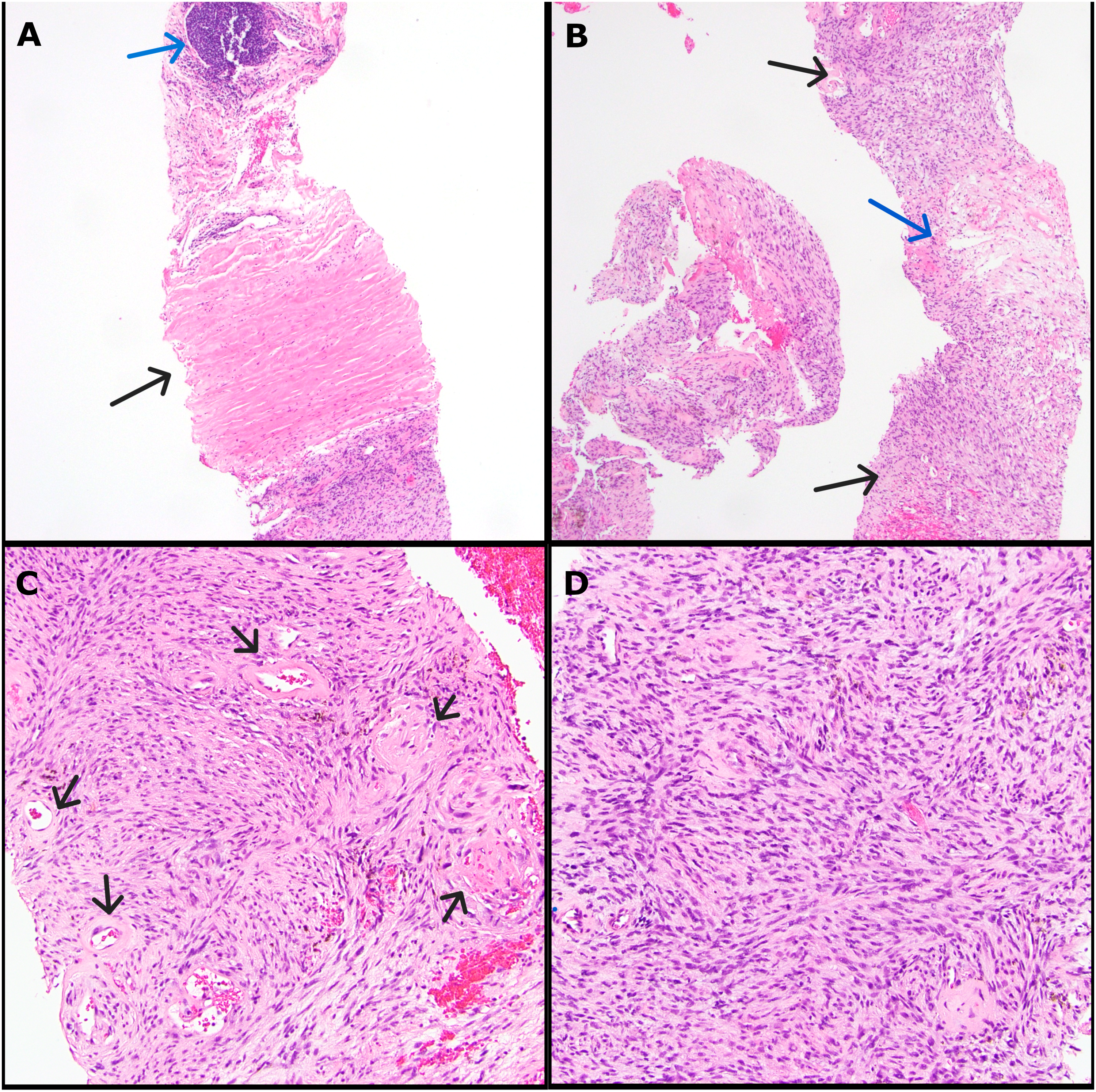
Histopathology of biopsy specimen. (A) 20× magnification showing tumor capsule (black arrow) and a pericapsular lymphoid aggregate (blue arrow); (B) 20× magnification with alternating hyper (black arrows) and hypocellular (blue arrow) areas; (C) 100× magnification with hyalinized vessels (black arrows); and (D) 200× magnification of hypercellular area with fascicles of spindle cells with bland, wavy nuclei.
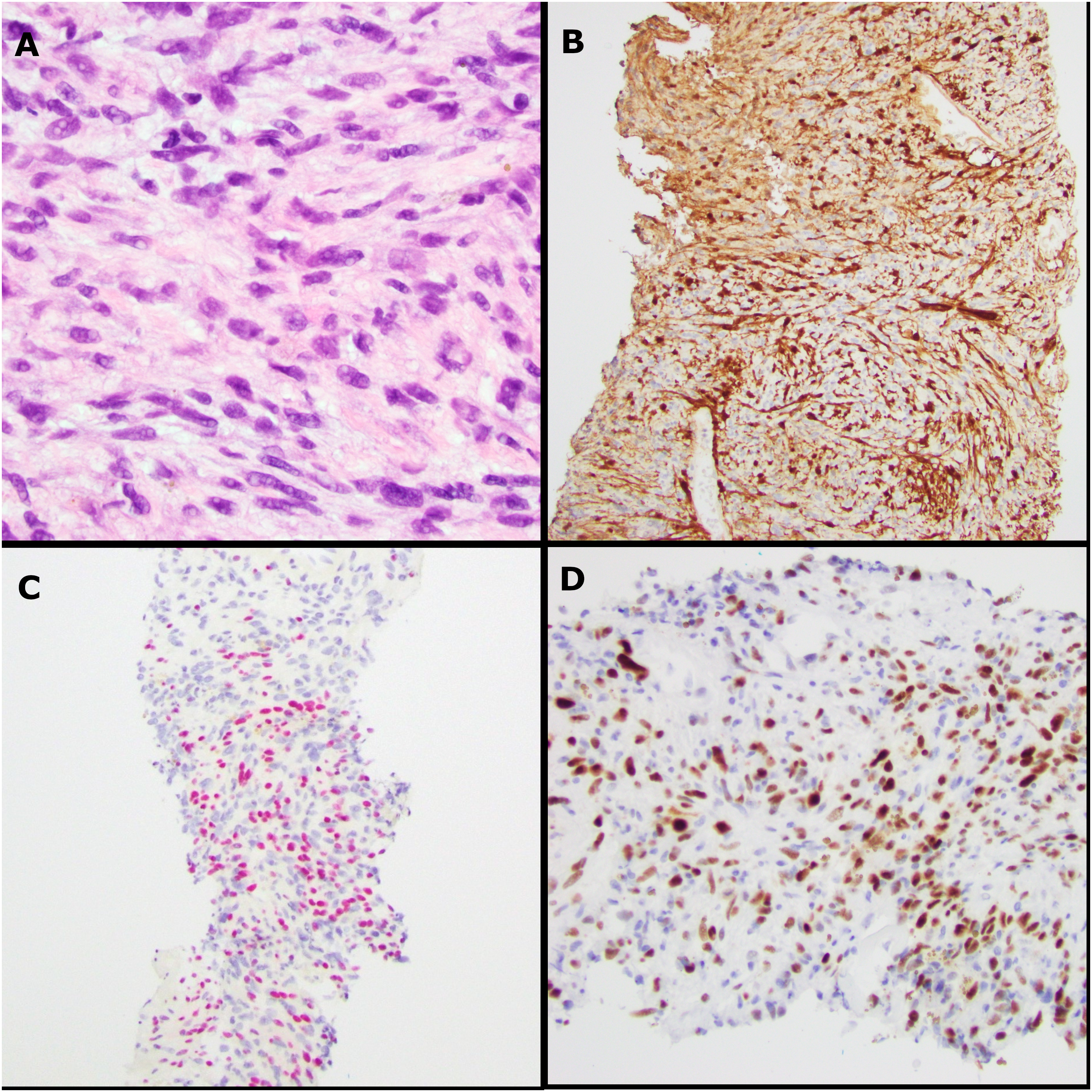
Histopathology and IHC of biopsy specimen. (A) 400× magnification of hypercellular area showing spindle cells with bland and wavy nuclei; (B) S100 IHC; (C) SOX10 IHC; and (D) p53 IHC. Abbreviation: IHC, immunohistochemistry.
The patient's clinical symptoms continued to worsen. She underwent multiple follow-up computed tomography (CT) scans over the next 6 months, which demonstrated a rapid increase in tumor size with the development of new soft tissue nodules along the lateral border of the psoas (Figure 1B, D, and E). Intraoperatively, the mass was fixed at the deeper aspect, intertwined with the motor nerves to the thigh muscles. Given her impaired motor nerve function, a decision was made to forgo further resection to preserve the remaining motor nerve function.
Macroscopically, the mass was 8.5 cm in the greatest dimension, with a rubbery, tan-white, and focally necrotic cut surface. Histological examination revealed a high-grade malignant neoplasm with sheets of plump spindle cells with moderate eosinophilic cytoplasm, pleomorphic nuclei with coarse chromatin, and prominent nucleoli (Figure 4A-C). Numerous mitotic figures, tumor necrosis, and vascular invasion were also identified. The separately submitted “tumor capsule” consisted of the same high-grade sarcoma and adjacent skeletal muscle without the residual capsule of the schwannoma. IHC showed that the tumor cells were negative for S100, SOX10, cytokeratin AE1/AE3, MDM2, CDK4, Melan-A (MART1), HMB45, alpha smooth muscle actin (SMA), desmin, MYOD1, myogenin, ERG, D2-40 (PDPN), DOG1, KIT, calretinin, WT1, SS18::SSX, and BRAF V600E. H3K27me3 and SMARCB1 IHC expression was retained. Next-generation sequencing (NGS) by the “FoundationOne Heme panel” revealed an NF2 V24_T28del variant at a variant allele frequency (VAF) of 47.3%. Mutations in TP53 (VAF 56.7%) and NRAS (VAF 20.2%), a subclonal RICTOR mutation (VAF 3.9%), and an equivocal EGFR amplification were also identified. The tumor mutational burden was 2 mutations per megabase. We retrospectively performed p53 IHC on both biopsy and excision specimens based on the NGS findings. We observed a mutant staining pattern, with 70% of the cells showing strong positivity in the core biopsy (Figure 3D), and >90% in the excised specimen (Figure 4D). The patient received 10 fractions of palliative radiotherapy. The disease progressed rapidly, and she died within a month of surgery.
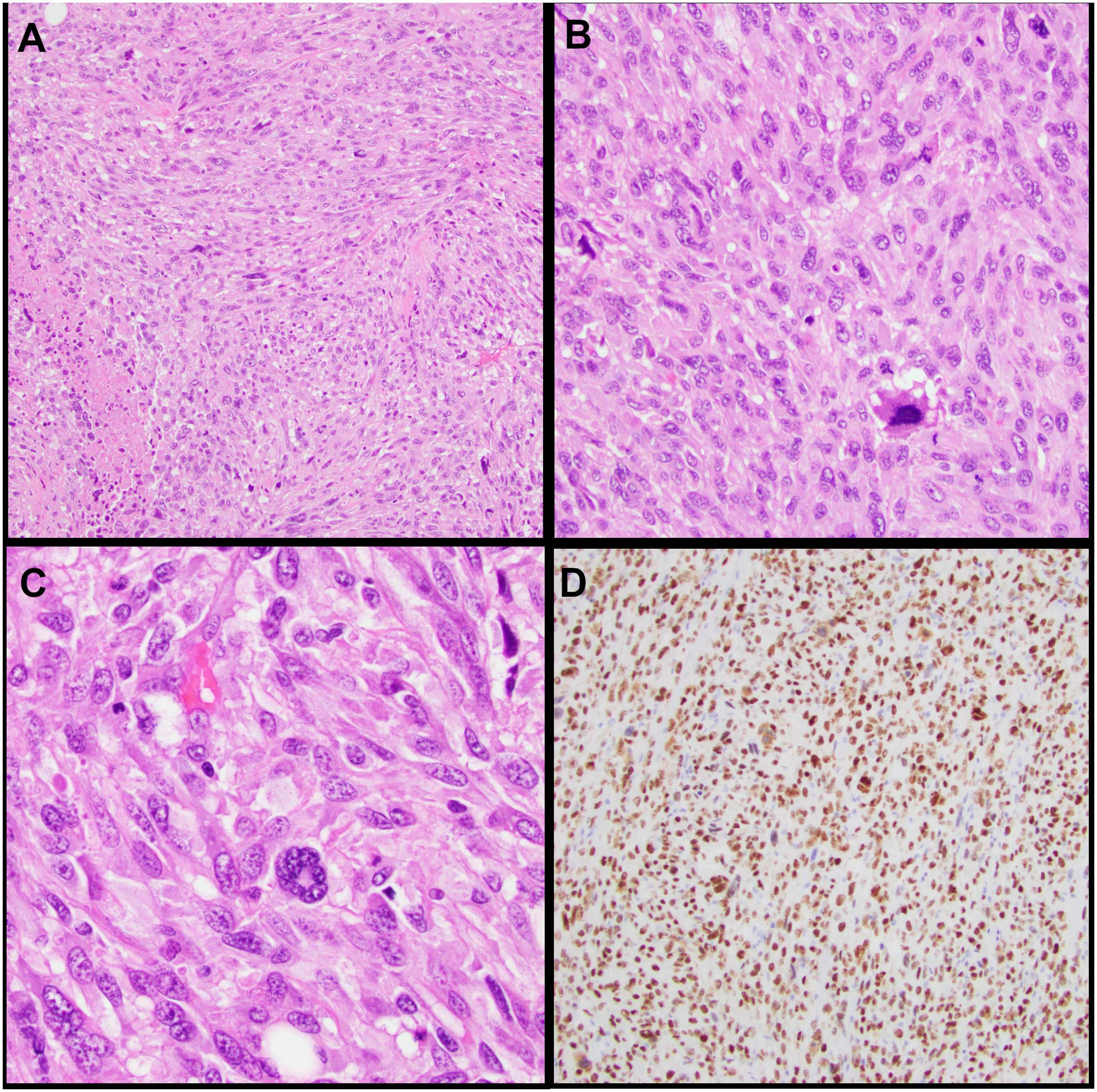
Histopathology of the excision specimen. (A) 100× magnification showing sheets of high-grade spindle cells with tumor necrosis; (B) 200× magnification with mitotic figures; (C) 400× magnification demonstrating the nuclear atypia of the spindle cells; and (D) p53 IHC. Abbreviation: IHC, immunohistochemistry.
Discussion
Even though our excision specimen was entirely replaced by a high-grade spindle cell sarcoma, including the separately examined pseudocapsule of the tumor, the prior biopsy served as evidence for a pre-existing schwannoma, thus fulfilling the criteria of Woodruff et al 9 for diagnosing malignant transformation of schwannoma. The finding of the NF2 V24_T28del frameshift mutation caused by a 15bp deletion in exon 1 of the NF2 gene on chromosome 22, with 47% VAF in the excision specimen, further supported that it arose from a schwannoma. We do not know the NF2 germline status of our patient; however, the penetrance rate of NF2 is nearly 100% by 60 years of age. 18 Our patient was 76 years old with a solitary soft tissue schwannoma. No bilateral vestibular schwannomas or other nervous system tumors were identified clinically, and she had no family history of schwannomas. However, given the morphology and the overall IHC pattern, her primary tumor was best considered a sporadic schwannoma.
The other SOX10-positive tumors with mild cytological atypia, which may be considered differential diagnoses for the biopsy of this tumor, include neurofibroma, granular cell tumor, soft tissue myoepithelioma, and metastatic melanoma. 19 However, these tumors are often not encapsulated and lack the hyalinized vessels typically seen in schwannomas. They feature more round or polygonal cells (granular cell tumor), may not be diffusely positive for SOX10/S100, and may be cytokeratin positive (soft tissue myoepithelioma). Overall, the NF2 mutation identified in the subsequent, overtly malignant form of this tumor ruled out the other possibilities.
The transformation of vestibular schwannomas into MPNSTs has been well-reported. However, a histopathological confirmation is only documented in 13 patients to date. 6 Some tumors are radiation associated, 8 and some are not.20,21 No association with “schwannomatoses” has been reported in these patients. Most of these tumors demonstrate a spindle cell morphology, with focal epithelioid cells in some. 21 Tumors with malignant transformation exhibit either focal,8,20,21 or complete loss of S100 expression. 22 Conversely, a histopathology-proven transformation of sporadic soft tissue schwannomas to MPNSTs has been reported in fewer than 30 patients, all of whom are summarized in Table 1.9–12,14–17 In most patients, the MPNST was of the epithelioid subtype9,10,12,16,17 (Table 1). A conventional spindle cell-type MPNST arising from a sporadic soft tissue schwannoma has only been reported in 2 patients11,14 (Table 1). The malignant component was identified within a benign schwannoma in all these tumors. Notably, in most patients, the malignant transformation occurred in deep-seated schwannomas.9,12,14,16,17 McMenamin et al reported 4 epithelioid MPNSTs arising within schwannomas. In one of these patients, the epithelioid MPNST presented as a rapid and painful enlargement of a long-standing neck lump postradiation. We excluded this patient from our literature review due to potential radiation association. Similar to our patient, malignant transformation presented as rapid and painful enlargement of a pre-existing lesion in two of their patients. 12 Our tumor is another example of a sporadic soft tissue schwannoma with malignant transformation to a conventional spindle cell-type MPNST.
Literature Review of Malignant Transformation of Schwannoma to MPNST.
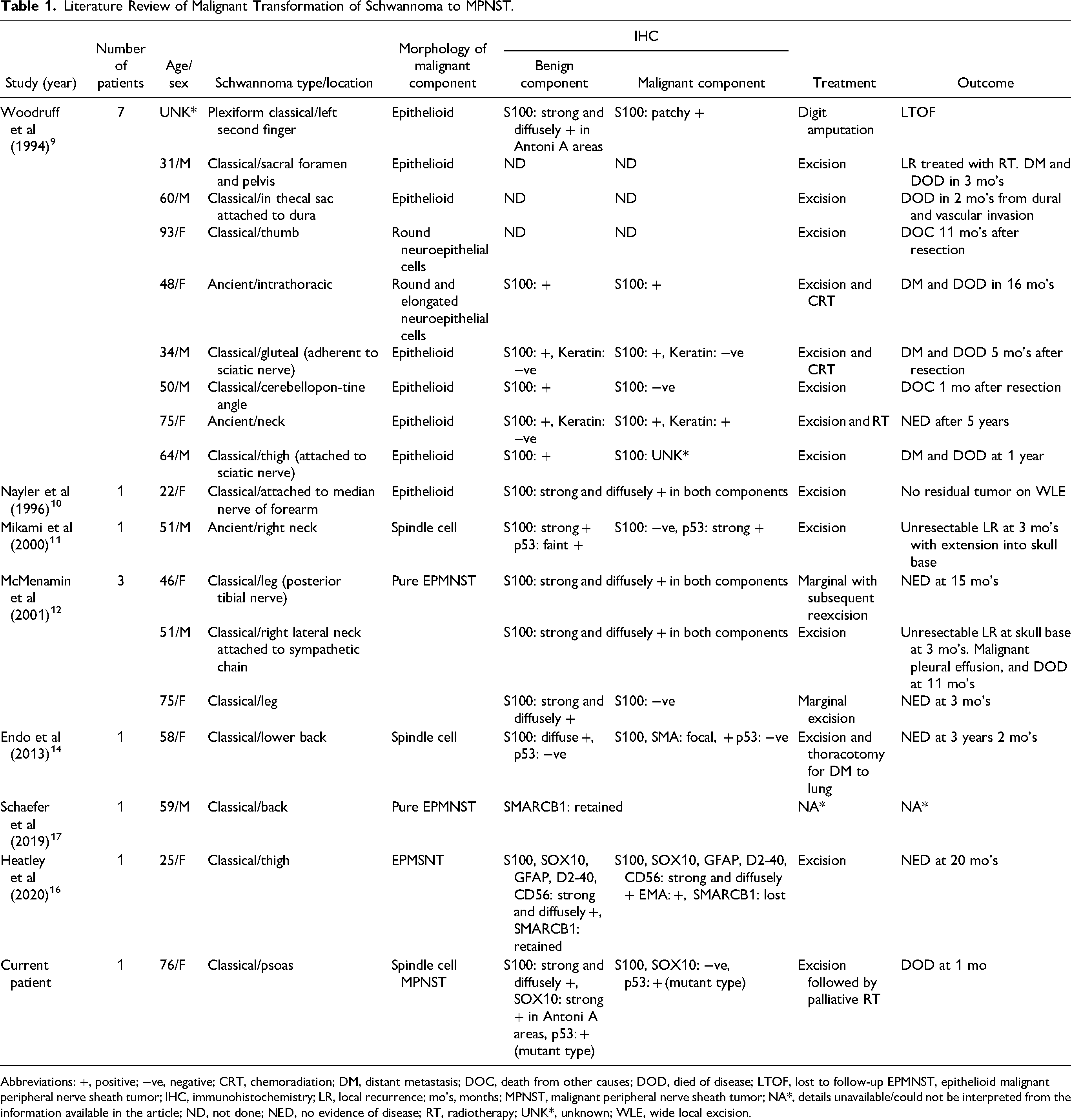
Abbreviations: +, positive; −ve, negative; CRT, chemoradiation; DM, distant metastasis; DOC, death from other causes; DOD, died of disease; LTOF, lost to follow-up EPMNST, epithelioid malignant peripheral nerve sheath tumor; IHC, immunohistochemistry; LR, local recurrence; mo's, months; MPNST, malignant peripheral nerve sheath tumor; NA*, details unavailable/could not be interpreted from the information available in the article; ND, not done; NED, no evidence of disease; RT, radiotherapy; UNK*, unknown; WLE, wide local excision.
The tumor suppressor TP53 is the most frequently mutated gene in cancer. In MPNSTs, the mutation frequency is variable, with a prevalence of 16.9%.23,24 However, when assessed through a TP53-associated gene signature, disruptions in the TP53 pathway, collectively termed the “TP53-mutated phenotype,” have been observed in up to 60% of MPNSTs. 24 Most TP53-mutated MPNSTs are of the conventional type and arise from neurofibromas. TP53 mutations in MPNST ex-schwannomas have only been documented in some case reports by IHC.7,11,25 Animal studies have provided evidence for a synergistic effect of NF2 and TP53 alterations in MPNST development. 26 Our tumor showed a G266R point mutation in the TP53 gene with a high VAF in the excised specimen. In retrospect, IHC staining showed mutant-type p53 staining in both excision and biopsy specimens. This indicated that the TP53 mutation occurred early in the malignant transformation process, even when the benign schwannoma morphology was maintained. This raises the question of whether p53 IHC should be considered in the biopsy work-up of schwannomas. Although we cannot draw a conclusion based on one patient, we would, in the future, consider p53 IHC in clinically suspicious tumors, such as worsening symptoms with rapidly enlarging or deep-seated tumors, as seen in our patient.
Other NGS findings in our tumor included NRAS p.Q61 K missense mutation (VAF 20.2%), which has been previously reported in 1 of 44 sporadic MPNSTs by Kao et al, 27 but has not been identified by genomic profiling of MSK-IMPACT, which included 50 MPNSTs in their cohort. 28 The missense mutation R89W in the RICTOR gene has a low VAF of 3.9%. Therefore, this could be subclonal and might be a source of tumor heterogeneity, but it is not a driver of tumorigenesis.
MPNST could be a diagnostic challenge for pathologists given its high-grade morphology and often negativity for S100 and SOX10 by IHC. Loss of H3K27me3 expression in MPNSTs corresponds to SUZ12 and EED gene mutations, and its identification has greatly improved the diagnostic accuracy of IHC. Although not completely specific for MPNST, it represents a valuable marker for this diagnosis. However, the frequency and extent of loss (partial vs complete) vary depending on the MPNST grade. 29 Our tumor is another example of retained H3K27me3 by IHC and correlates with the absence of SUZ12 and EED gene mutations by NGS. Based on morphology and lack of lineage-specific markers, the excised high-grade sarcoma could have been classified as an undifferentiated pleomorphic sarcoma in the absence of prior core biopsy or NGS data. This case raises the possibility that some tumors labeled as undifferentiated pleomorphic sarcomas may actually represent MPNSTs.
Conclusion
Herein, we report a novel scenario of a sporadic soft tissue schwannoma in an older woman that rapidly transformed into a spindle cell-type MPNST with an early TP53 mutation, NF2 loss, NRAS, and subclonal RICTOR gene mutations. A unique feature of this tumor is the mutant-type p53 expression detected by IHC in a morphologically benign schwannoma. This raised the practical question of whether we should consider p53 IHC on biopsies of schwannomas, particularly those in deep-seated locations. The resected tumor histologically resembled the undifferentiated pleomorphic sarcoma with no immunoreactivity for any lineage-specific markers, which also raised the speculation that some of the so-called undifferentiated pleomorphic sarcomas might be MPNSTs.
Footnotes
Author Contributions
XW developed the idea and concept, while MR prepared the manuscript. VU, DH, and AH guided MR in drafting the manuscript, offering expert feedback and direction. GD contributed specialized expertise in the interpretation of radiology images.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Ethical Approval
Not applicable, because this article does not contain any studies with human or animal subjects. The study is retrospective, and there is no new operation.
Informed Consent
Not applicable, because this article does not contain any studies with human or animal subjects.
Trial Registration
Not applicable, because this article does not contain any clinical trials.