
Abstract
Secondary polycythemia is a paraneoplastic syndrome observed in tumors with excessive erythropoietin (EPO) production. Renal cell carcinoma (RCC) and cerebellar hemangioblastoma are the 2 most well-known tumors to induce secondary polycythemia. Hemangioblastomas occurring in the kidney are rare. In this work we present a case of renal hemangioblastoma that caused erythrocytosis in a 19-year-old man. We demonstrated intratumoural EPO production by immunohistochemistry, and conducted whole-exome sequencing to evaluate possible genetic alterations that reported to induce tumor-related polycythemia. In spite of an indolent clinical behavior, renal hemangioblastoma is difficult to differentiate from RCC not only clinically, but also histopathologically. Given that RCC is the most well-known renal tumor to induce erythrocytosis, the uncommon manifestation of polycythemia in renal hemangioblastoma, as shown in our case, can cause further diagnostic challenges. Renal hemangioblastoma should be listed in the differential diagnoses of renal tumors presenting with erythrocytosis, apart from the most common RCC.
Introduction
Secondary polycythemia is a paraneoplastic syndrome observed in tumors with excessive erythropoietin (EPO) production. Up to one-third of cases of tumor-associated polycythemia are caused by renal tumors, most of which are renal cell carcinomas (RCC).1,2 While cerebellar hemangioblastoma is also one of the most well-known tumors to cause secondary polycythemia, hemangioblastomas outside the central nervous system (CNS) are rarely reported to be associated with erythrocytosis. Herein, we present a case of renal hemangioblastoma manifesting with polycythemia.
Case Presentation
The patient was a 19-year-old man with no systemic disease. During a hospital visit for abdominal pain, sonography and a subsequent computed tomography (CT) scan demonstrated a mass in his right kidney [Figure 1(A)]. Laboratory tests revealed polycythemia (with hemoglobin up to 20.8 g/dL and hematocrit up to 65.8%). He had neither familial history nor clinical evidence of von Hippel-Lindau (VHL) disease. Partial nephrectomy was performed following needle biopsy, which showed a benign vascular lesion. The postoperative course was smooth, and his hemoglobin and hematocrit levels were reduced to be within normal ranges [Figure 1(B) and (C)]. A CT scan noted no recurrence 10 months after surgery.
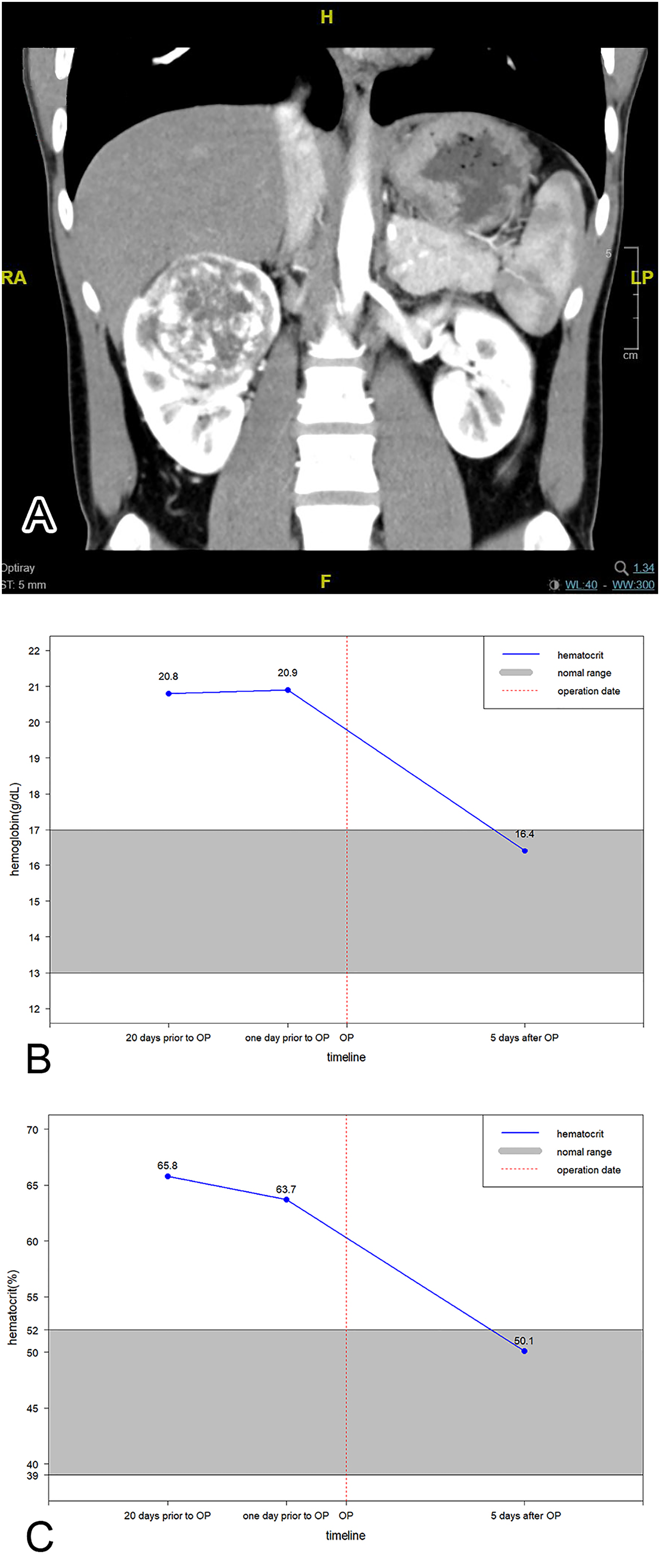
(A) CT scan showing a well-circumscribed mass in the upper pole of the right kidney. (B) and (C) The patient's erythrocytosis resolved following surgery. The normal ranges of hemoglobin and hematocrit are 13.0 to 17.0 g/dL and 39.0% to 52.0%, respectively. Abbreviation: CT, computed tomography.
The partially nephrectomized kidney measured 5.7 × 4.6 × 3.9 cm in size. A brown and solid tumor measuring 4.1 × 3.5 × 2.8 cm in size was found [Figure 2(A)]. Microscopically, the tumor consisted of aggregates of ovoid cells resting on a rich vascular network. These ovoid cells had clear cytoplasm containing small vacuoles [Figure 2(B)-(D)]. Mitotic figures and tumor necrosis were not observed. Immunohistochemically, the tumor cells were positive for S100 [Figure 3(A)], inhibin [Figure 3(B)], and PAX8, while negative for pan-keratin, HMB-45, melan-A, and chromogranin A. Additionally, EPO expression was observed in the majority of tumor cells [Figure 3(C)]. CD31 [Figure 3(D)] stain highlighted the vascular network but not the tumor cells. These findings supported the diagnosis of hemangioblastoma of the kidney. Genetic mutations such as VHL, EPAS1, EGLN1/2, and ACO1 were not detected by whole-exome sequencing (WES), and no significant deletions were detected by copy number variation analysis.
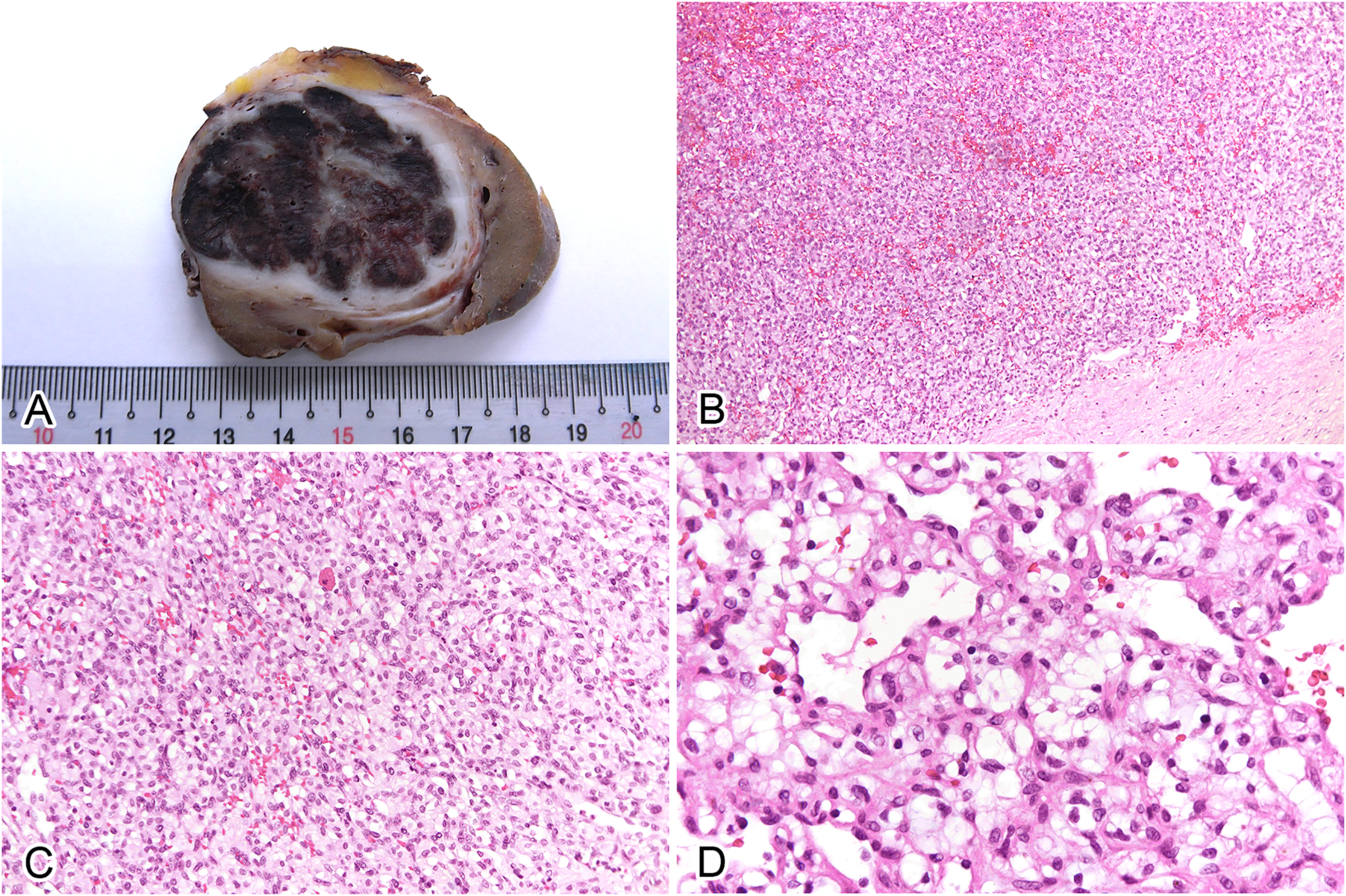
(A) Gross appearance of the tumor showing a brown and solid mass. (B) The tumor is well-defined and vascular rich (H&E, × 100). (C) The tumor cells are arranged in a sheet-like pattern. They are ovoid with clear to pale eosinophilic cytoplasm (H&E, × 200). (D) Small microvacuoles can be seen in the cytoplasm of the stromal tumor cells (H&E, × 400). Abbreviation: H&E, hematoxylin and eosin.
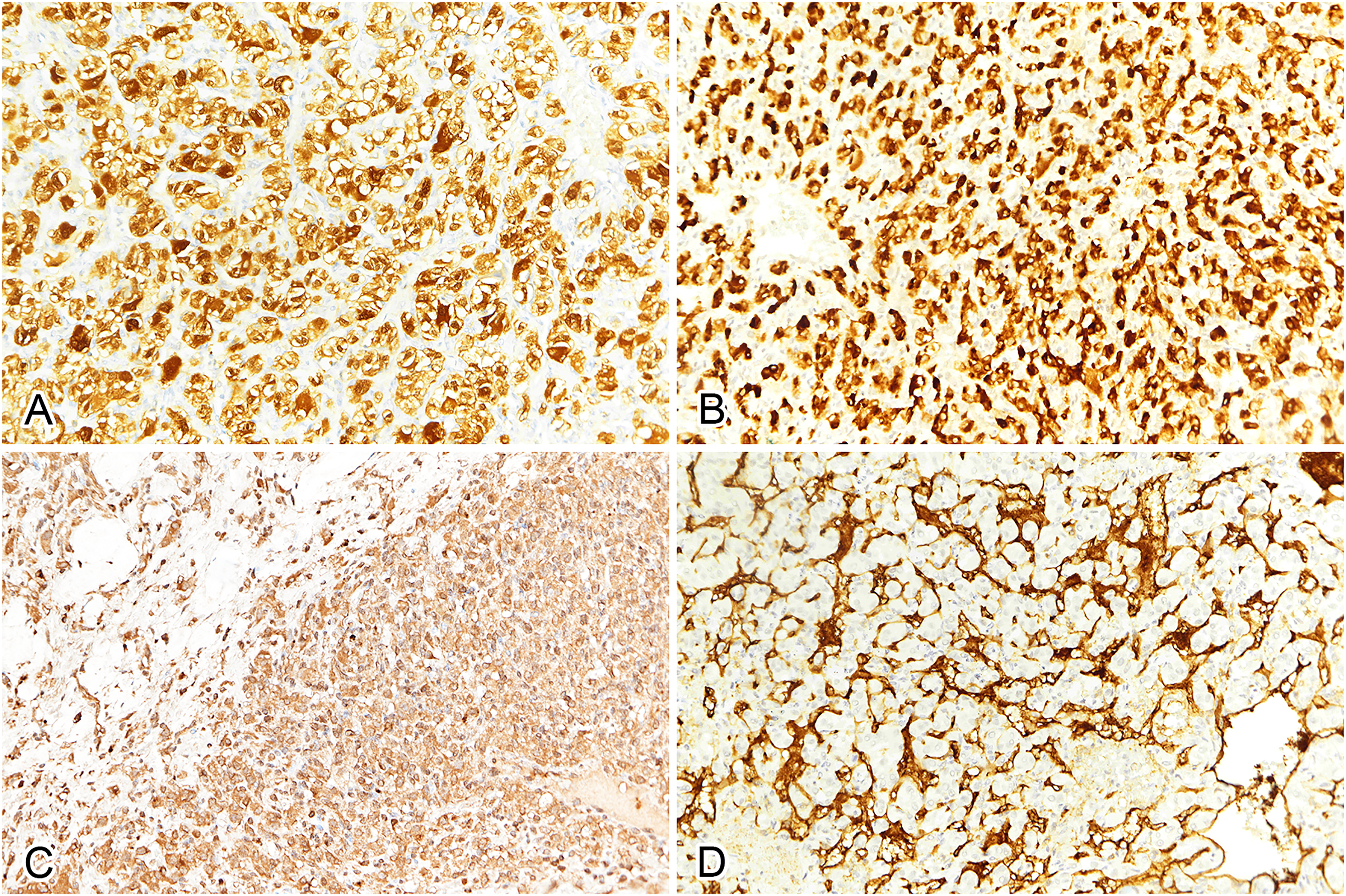
The tumor cells are immunopositive for (A) S100 (IHC, × 200), (B) inhibin (IHC, × 200), and (C) EPO (IHC, × 200). (D) CD31 highlights the rich vascular network (IHC, × 200). Abbreviations: EPO, erythropoietin; IHC, immunohistochemistry.
Discussion
Primary polycythemia results from the overproduction of red blood cells caused by abnormalities in hematopoietic progenitors. In contrast, secondary polycythemia results from conditions associated with EPO production, such as neoplasms. Tumors known to cause erythrocytosis include cerebellar hemangioblastoma, pheochromocytoma, hepatocellular carcinoma (HCC), and renal tumors such as RCC and Wilms tumor. 1
Among these tumors, cerebellar hemangioblastoma and clear cell renal cell carcinoma are well-known manifestations of VHL disease, an autosomal-dominant disorder caused by pathogenic germline mutations of the VHL gene. Loss of VHL protein function leads to the accumulation of HIF-α, which in turn transactivates downstream hypoxia-inducible molecules that can promote angiogenesis and tumor progression. EPO is one of these molecules, and overexpression of EPO in cerebellar hemangioblastoma and clear cell RCC has been reported in many studies.3,4
Apart from VHL, there are other genes reported to cause tumor-related polycythemia, including (1) EPAS1 mutation in HCC, paraganglioma, and somatostatinoma5,6; (2) EGLN1/2 mutations in pheochromocytoma/paraganglioma 7 ; (3) ACO1 mutation in pheochromocytoma. 8 All of these genetic mutations influence the hypoxia signaling pathway, either directly or indirectly. In addition, Ke et al recently identified that mitochondrial DNA mutations and subsequently impaired respiratory metabolism inhibit PHD activity and stabilize HIF-α, leading to increased EPO expression in HCC with erythrocytosis. 9
Hemangioblastomas typically occur in the cerebellum. Cerebellar hemangioblastoma can be either sporadic (57%-75%) or associated with VHL disease (20%-43%). 10 Inactivation of the VHL gene can be detected in 78% of sporadic cerebellar hemangioblastomas. 11 While symptoms are generally caused by the mass effect of the lesion, secondary polycythemia resulting from EPO production is well documented. 4 Morphologically it is characterized by a prominent capillary network and stromal cells with clear to eosinophilic cytoplasm containing intracytoplasmic microvacuoles. Nuclear pleomorphism is usually minimal, but can be obvious in some cases. 12 Mitotic activity is generally low, and necrosis is not observed. Immunohistochemically the neoplastic cells express inhibin, NSE, and S100, but not endothelium-associated markers. SMA and focal EMA expression in some cases have also been reported, causing further possible diagnostic pitfall. 12 The prognosis is excellent after complete excision. 13
Hemangioblastomas rarely develop in the kidney. To date, <40 cases have been reported in the English literature.12,14–24 In spite of bearing similar morphological features, renal hemangioblastoma differs from its cerebellar counterpart in several aspects. Unlike cerebellar hemangioblastomas, renal hemangioblastomas are not associated with VHL disease, and no VHL gene mutation, loss of heterozygosity of chromosome 3p, or chromosome 3p deletion has been detected in renal hemangioblastoma in various studies.15–21 Brachyury expression is not observed in extraneural hemangioblastomas. The stromal cells in renal hemangioblastoma are immunopositive for PAX8, while those in cerebellar hemangioblastoma are not. 14 The endothelial cells in cerebellar hemangioblastoma typically express GLUT1, as CNS endothelial cells do; the endothelial cells in renal hemangioblastoma, on the other hand, do not express GLUT1, similar to normal peripheral endothelium. These differences in immunophenotypes support the theory that tumor cells of hemangioblastomas express site-specific antigens, possibly due to derivation from organ-specific pluripotent cells. 14
The primary differential diagnosis of renal hemangioblastoma is clear cell RCC. Due to the low occurrence rate and lack of specific clinical/radiological findings, renal hemangioblastoma is usually preoperatively diagnosed as RCC. Approximately one-half of renal hemangioblastoma cases in the literature underwent radical nephrectomy, mainly under the clinical impression of RCC.12,14–24 Given that RCC is the most well-known renal tumor to induce erythrocytosis, the uncommon manifestation of polycythemia in renal hemangioblastoma can cause further diagnostic challenges. Histopathologically, renal hemangioblastoma is also reminiscent of clear cell RCC. Both tumors are characterized by a rich vascular network, ovoid cells with a clear-to-eosinophilic cytoplasm, and PAX8 expression. Presence of intracytoplasmic lipid vacuoles, expression of inhibin and S100, and negativity for epithelial markers such as cytokeratin are clues for diagnosing hemangioblastoma.
There are also other histological mimickers of renal hemangioblastoma. The clear and vacuolated cytoplasm of the stromal tumor cells may resemble lipomatous tumors and fat-forming solitary fibrous tumor. However, true lipoblasts with scalloped nuclei and fat formation are not seen in hemangioblastoma. CDK4 and MDM2 stains can be of use in excluding well-differentiated liposarcoma. Abundant mitotic figures and necrosis, while frequently seen in pleomorphic liposarcoma, are not features of hemangioblastoma. Paraganglioma and epithelioid angiomyolipoma are also composed of cells with clear or eosinophilic cytoplasm, but bubbly lipid microvacuolization are not observed. Paraganglioma is characterized by a “zellballen” architectural pattern, in contrast with the sheet-like pattern of hemangioblastoma. An appropriate immunohistochemistry panel can also be helpful: diffuse synaptophysin and chromogranin expression indicates paraganglioma, while HMB-45 and melan-A expression is characteristic of epithelioid angiomyolipoma.
The phenomenon of renal hemangioblastoma-induced polycythemia was reported only once in a patient without evidence of VHL disease. 16 Here, we present an additional case and document intratumoral EPO expression by immunohistochemistry. We also conducted WES to evaluate possible genetic alterations reported in the literature. Genetic mutations in VHL, EPAS1, EGLN1/2, and ACO1 were not found in our case. However, mitochondrial DNA was not tested.
In conclusion, renal hemangioblastoma is a rare tumor that can cause secondary polycythemia. It should be listed in the differential diagnoses of renal tumors presenting with erythrocytosis, apart from the most common RCC. In contrast to RCC, renal hemangioblastoma is an indolent tumor. Pre-operative needle biopsy, as shown in our case, can help identify the tumor's benign nature and indicate nephron-sparing surgery instead of radical nephrectomy.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Ethical Approval
Not applicable, because this article does not contain any studies with human or animal subjects.
Informed Consent
Not applicable, because this article does not contain any studies with human or animal subjects.
Trial Registration
Not applicable, because this article does not contain any clinical trials.