
Abstract
Thymomas are tumors of the mediastinum often associated with autoimmune conditions, in particular myasthenia gravis. In contrast, among the fewer than 40 reports of metaplastic thymoma, myasthenia gravis is rarely found. We describe the fourth patient, and first man, with metaplastic thymoma and myasthenia gravis. A 34-year-old had acute onset of double vision with associated dysphagia and was found to have an elevation of serum acetylcholine receptor antibodies. He underwent a transsternal thymectomy. Tissue sections showed a biphasic proliferation of keratin-positive epithelial cells with a complement of spindle cells confirming the diagnosis of metaplastic thymoma. Terminal deoxynucleotidyl transferase (TDT)-positive T lymphocytes were rare and only found in the periphery of the tumor, consistent with thymic remnant. A YAP1::MAML2 gene fusion, with an in-frame fusion between genes YAP1 Exon5 (NM_001130145) and MAML2 Exon2 (NM_032427) was found, supporting further the diagnosis of metaplastic thymoma (Anchored multiplex RNA sequencing [Archer Dx, Boulder, CO] assay). The patient's gender and relatively young age, the presence of an autoimmune condition, and the lack of lymphocytic infiltrate all contribute unusual features to this case and suggest avenues for further exploration.
Introduction
Thymomas are uncommon tumors of the mediastinum and can be detected incidentally or associated with myasthenia gravis and other autoimmune diseases in 30% to 40% of patients.1–3 Among the types of thymomas described by the current WHO classification of tumors (5th ed.), metaplastic thymoma is the rarest with fewer than 40 patients reported in the English-language literature. 4 In many cases they are asymptomatic and found incidentally during imaging for other indications. Metaplastic thymoma in association with autoimmune diseases has been rarely reported. Here we describe a patient with metaplastic thymoma presenting with myasthenia gravis, further notable for the patient's young age, male gender, and the presence of the autoimmune condition. We discuss the literature and explore new advances in genomic analysis of this tumor and understanding of the relationship between thymoma and myasthenia gravis.
Clinical Presentation
A 35-year-old man with a medical history of hypertension, seasonal allergies, nasal polyposis, and a distant history of smoking noticed recurrent generalized fatigue and physical activity intolerance over a period of 6 to 7 months, accompanied by occasional choking episodes and shortness of breath. The patient developed acute right-sided ptosis, worsening double vision, and worsening dysphagia. He was evaluated by an ophthalmologist after reporting he felt he could not open his eyes or see clearly to drive.
A brain MRI was normal. A chest CT revealed a 7.2 × 5.4 × 5.0 cm mediastinal mass (Figure 1). PET/CT identified a lobulated predominantly soft tissue mass in the left anterior mediastinum with homogeneous avid FDG uptake (SUV 13.2). There was no evidence of local invasion or metastatic disease. An acetylcholine receptor (AChR) antibody concentration of 7.01 mmol/L confirmed a diagnosis of myasthenia gravis.
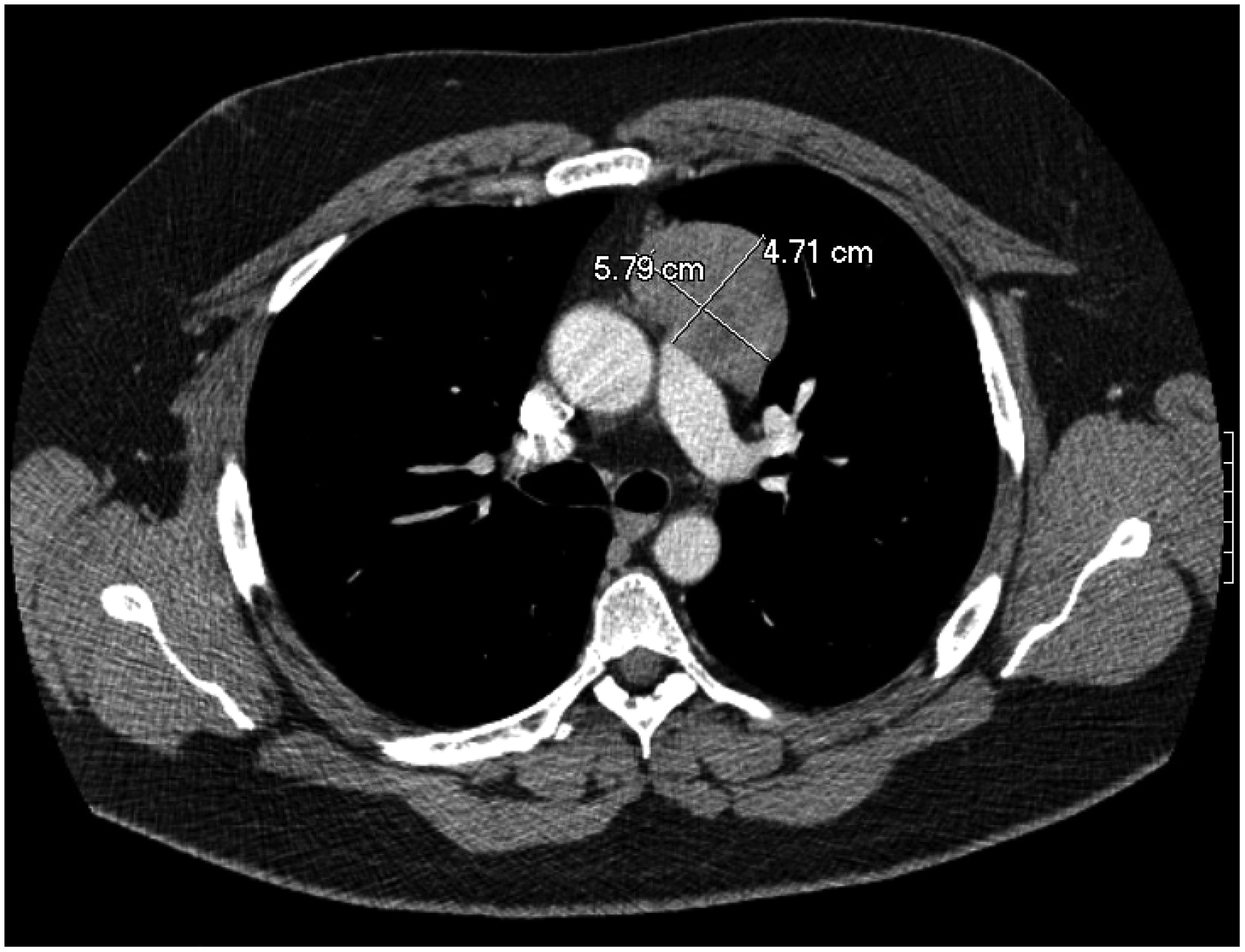
Axial view of mediastinal mass.
Pre-thymectomy treatment consisted of pyridostigmine 60 mg TID and prednisone titrated up over 6 weeks to 60 mg daily. Dysphagia, generalized fatigue, bilateral ptosis, and general function began to improve after 1 week of treatment.
Surgical Treatment
After prednisone was reduced to 30 mg daily, the patient underwent an attempted left robotic-assisted thoracoscopic thymectomy. However, given the patient's body habitus and limited intraoperative visualization, the procedure was converted to a trans-sternal extended thymectomy. A 13 cm mass was resected en bloc with the thymus and perithymic fat. There was no invasion of surrounding structures. The patient had an uneventful postoperative course, and he was discharged home on the second postoperative day.
Pathology Findings
Grossly the patient's tumor measured 8.0 × 7.0 × 4.0 cm. The cut surface was tan-white, firm, and multilobulated. On microscopic examination, tissue sections showed a lymphocyte-poor biphasic proliferation of epithelial cells with solid and trabecular pattern and spindle cells with fascicular and storiform growth (Figures 2 and 3). The epithelial cells were small- to medium-sized, with moderate amounts of eosinophilic cytoplasm and round to oval nuclei with vesicular chromatin. The spindle cells were medium-sized with elongated cytoplasm with fusiform nuclei. A subset of the tumor cells showed large nuclei with pseudoinclusions. There was no increased mitotic activity. Patchy areas of infarct-type necrosis were seen, but no true tumor necrosis was observed. The background showed involuted thymic tissue and the tumor did not invade into the capsule or vessels. Immunohistochemical stains showed that the tumor cells were positive for keratin AE1/AE3, KRT5/6, and p40, while they were negative for CD5 and KIT (Figure 4). No significant population of lymphoid cells positive for terminal deoxynucleotidyl transferase (TDT), also known as DNA nucleotidylexotransferase (DNTT), were seen within the tumor (Figure 5).
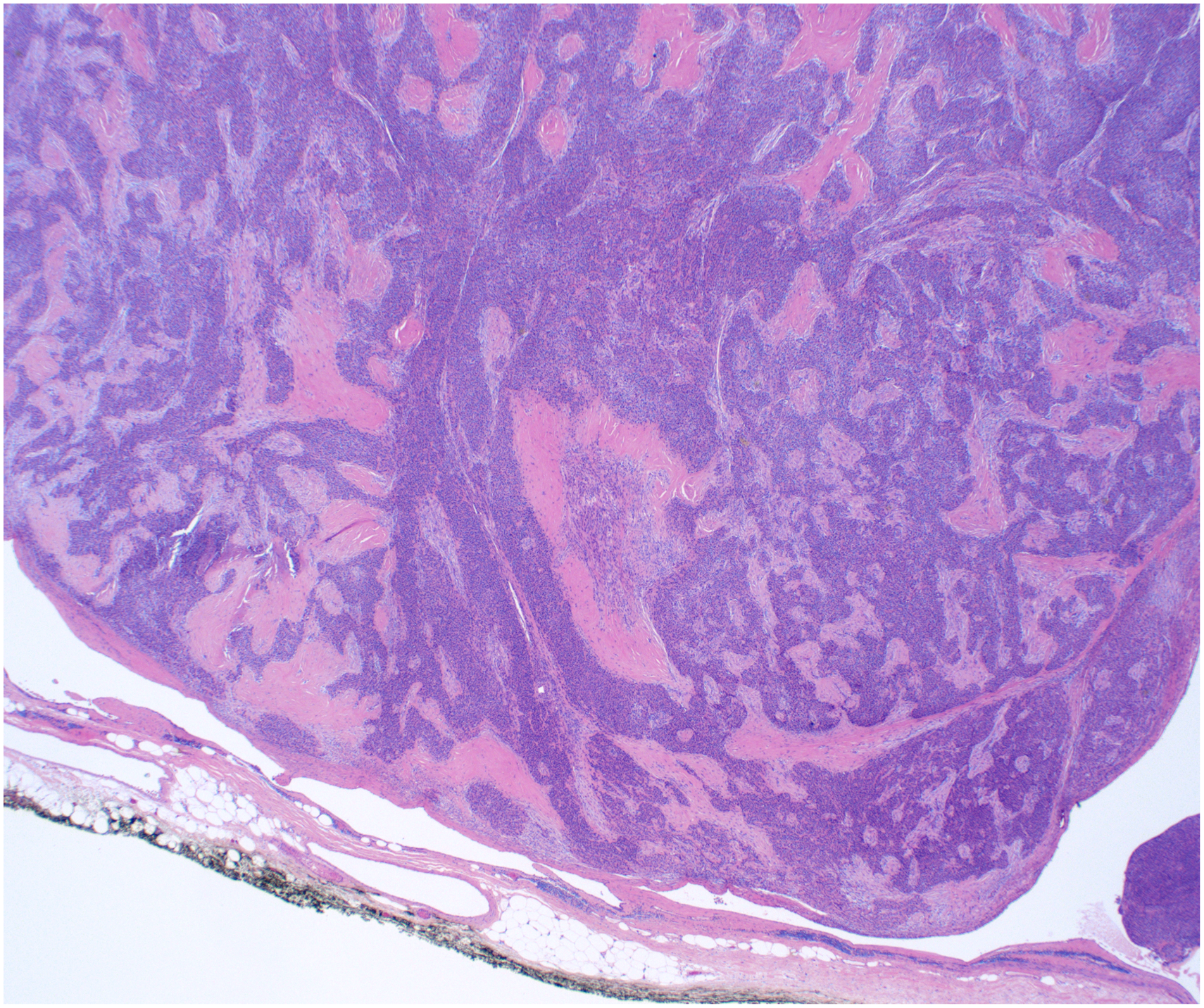
H&E stain, low power view 2×. Biphasic proliferation of epithelial cells and spindle cells.
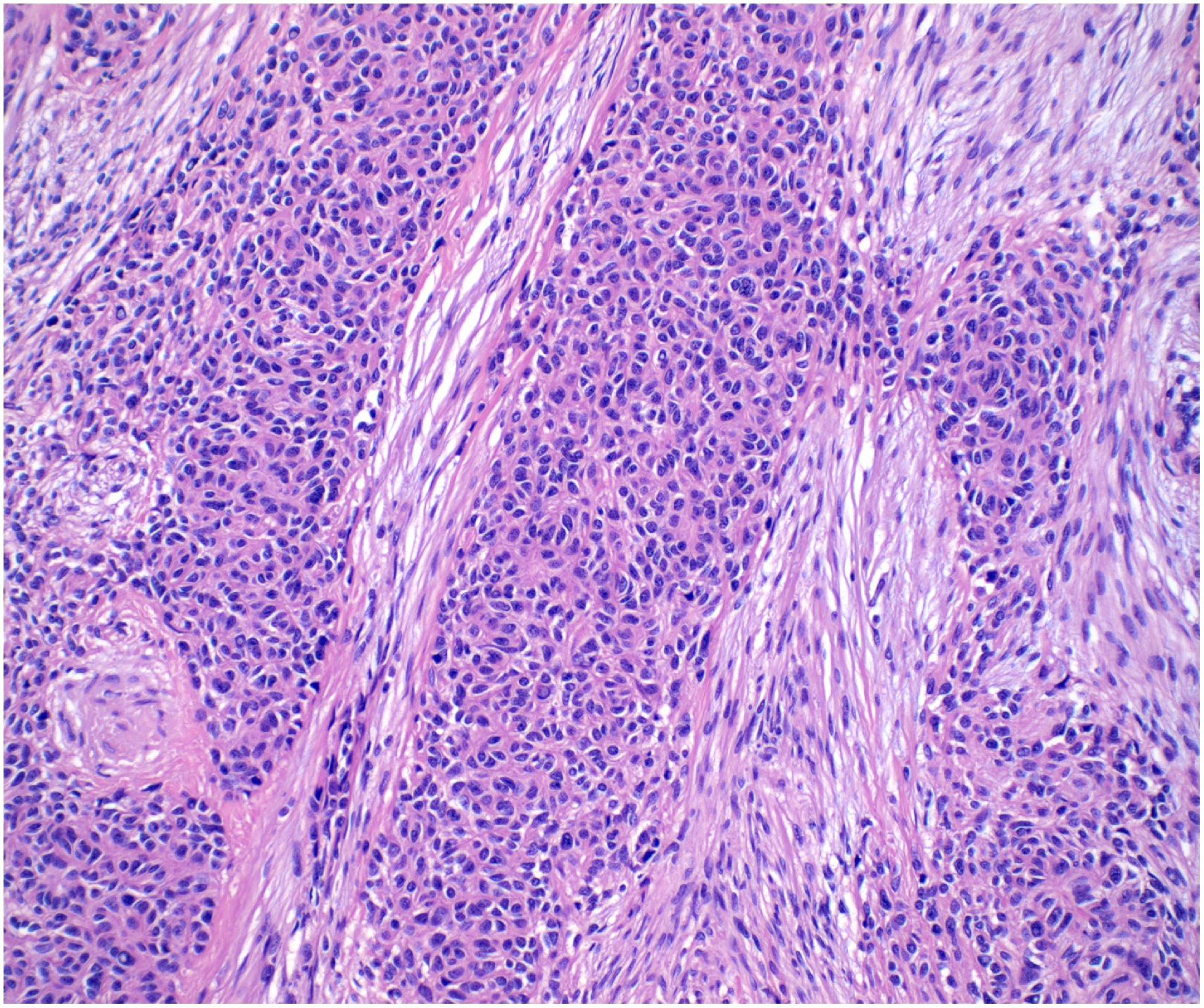
H&E stain, high power view 20×. Biphasic proliferation of epithelial cells and spindle cells.
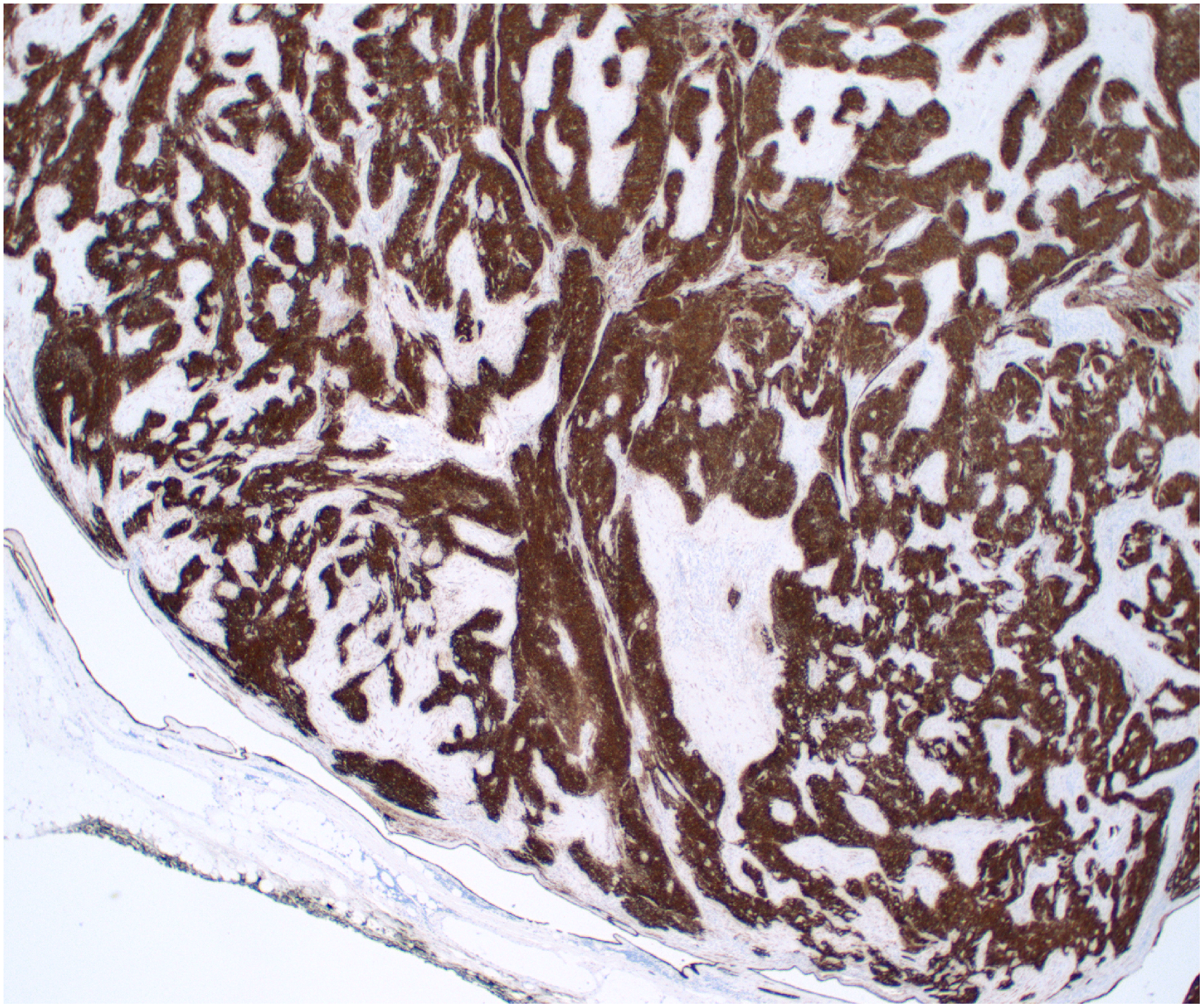
Keratin AE1/AE3 stain, 2×. Tumor cells were positive for keratin AE1/AE3, KRT5/6, and p40, while they were negative for CD5 and KIT.
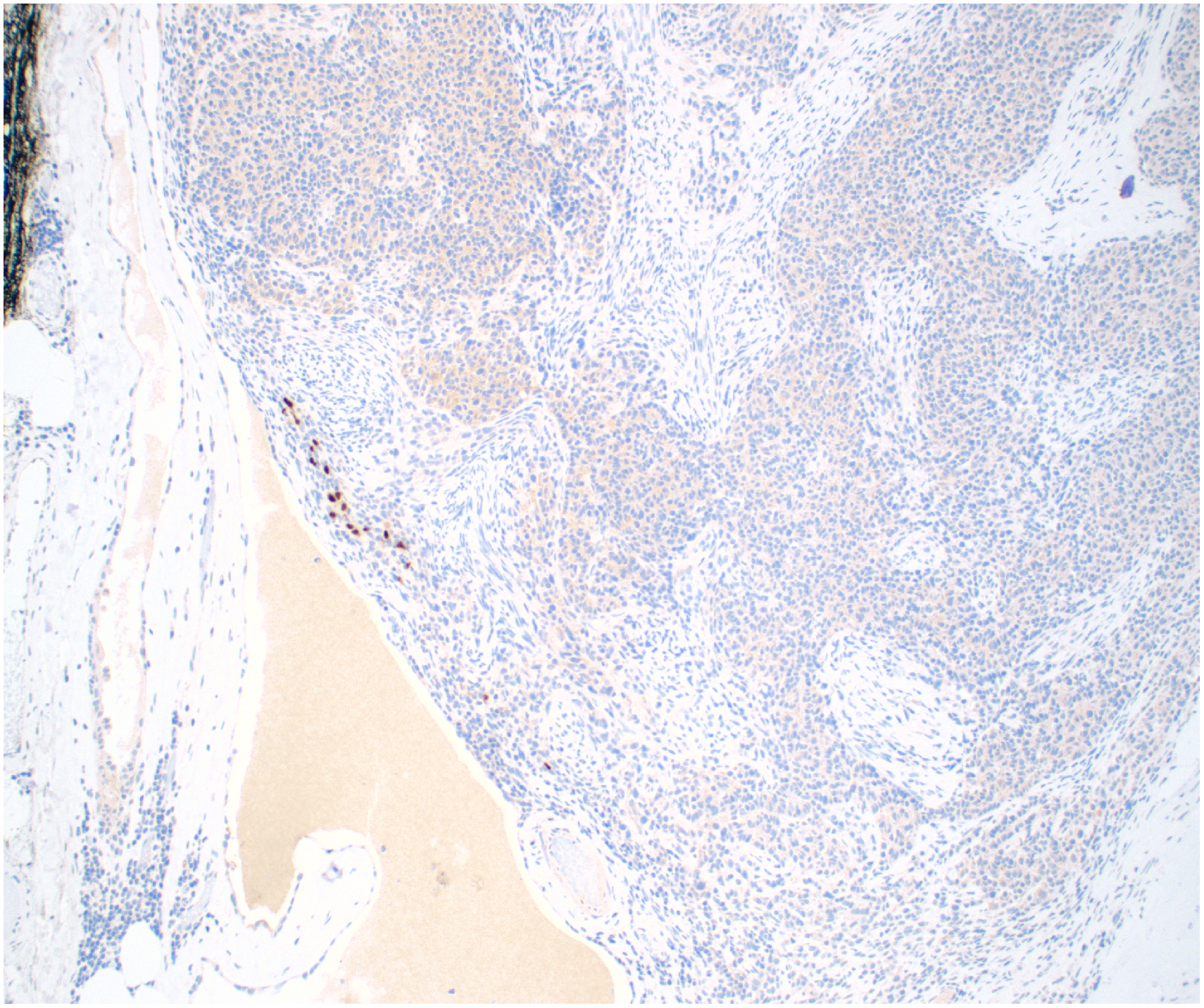
TDT stain, 4×. No significant population of TDT-positive lymphoid cells was observed.
A diagnosis of a well-differentiated squamous cell carcinoma was considered, however the lack of at least moderate cytologic atypia and mitotic activity spoke against this diagnosis. The absence of lymphocytes made thymoma AB and B3 unlikely. Based on the histology described above, a diagnosis of metaplastic thymoma was rendered.
Molecular Findings: Anchored Multiplex RNA Sequencing
Anchored multiplex RNA sequencing (Archer Dx, Boulder, CO) assay was performed on sections obtained from formalin-fixed paraffin embedded tissue. The detailed procedure has been previously described. Unidirectional gene-specific primers were designed to target specific exons in 123 genes known to be involved in oncogenic fusions in solid tumors. In brief, RNA was extracted from formalin-fixed paraffin embedded specimens, followed by complementary DNA synthesis and library preparation. Anchored multiplex polymerase chain reaction amplicons were sequenced on an Illumina Miseq sequencer (Illumina, San Diego, CA), and the data were analyzed by using Archer software (ArcherDX, Boulder, CO).
A YAP1::MAML2 gene fusion, with an in-frame fusion between genes YAP1 Exon5 (NM_001130145) and MAML2 Exon2 (NM_032427) was found, supporting the diagnosis of metaplastic thymoma.
Clinical Follow-up
Six months after surgery the patient continues to be symptomatic requiring prednisone, intravenous immunoglobin, and tacrolimus.
Discussion
Metaplastic thymomas are the rarest form of thymomas. Originally described by Yoneda et al in 1999, fewer than 40 cases have been described since in the English-language literature.5–13 An earlier report of 6 cases was presented by Suster et al in 1997, although the term “metaplastic thymoma” was not yet in use. 14 Most cases reported occur in Asia. The age distribution of the patients with metaplastic thymoma ranges from 29 to 71 with a median of 53 years. There is a slight female predilection (M:F ratio: 1:1.5). Clinically, only 3 patients were described to have symptoms of myasthenia gravis. Most of the patients had an excellent outcome, except for those tumors demonstrating sarcomatous component or transformation.7,9,15
Previously described as thymoma with pseudosarcomatous stroma, low-grade metaplastic carcinoma, biphasic thymoma, mixed polygonal and spindle cell type, the WHO classification of thoracic tumors recognized metaplastic thymoma as its own entity in 2004. 4 The diagnosis is based on histologic findings of a biphasic tumor with islands of polygonal thymic epithelial cells separated by abundant spindle cells with no cytologic atypia. The epithelial component may display mild atypia, but there is no mitotic activity. The tumor is typically encapsulated and well circumscribed. Unless there is sarcomatoid transformation, as it has been described in 3 cases previously mentioned, there is no cytologic atypia, increased mitotic activity, or areas of necrosis.7,9,15
The origin of the spindle cells is unclear, and whether they are simply stromal cells or the result of a mesenchymal transformation of the epithelioid tumor cells is debated. In 2012, Biao Liu et al considered the spindle cell component to be a mesenchymal metaplasia of tumor cells through a metaplastic phenomenon known as epithelial-mesenchymal transition (EMT). 11 In their study of 7 patients, the spindle cell component lost expression of E-cadherin, yet the epithelial cells were diffusely and homogeneously positive for this molecule. In the EMT hypothesis, the transdifferentiated spindle component almost always displays the loss of keratin and E-cadherin expression and up-regulates expression of mesenchymal markers including vimentin (Figure 6). Our patient displays loss of E-cadherin in the spindle cells, consistent with observations of others.
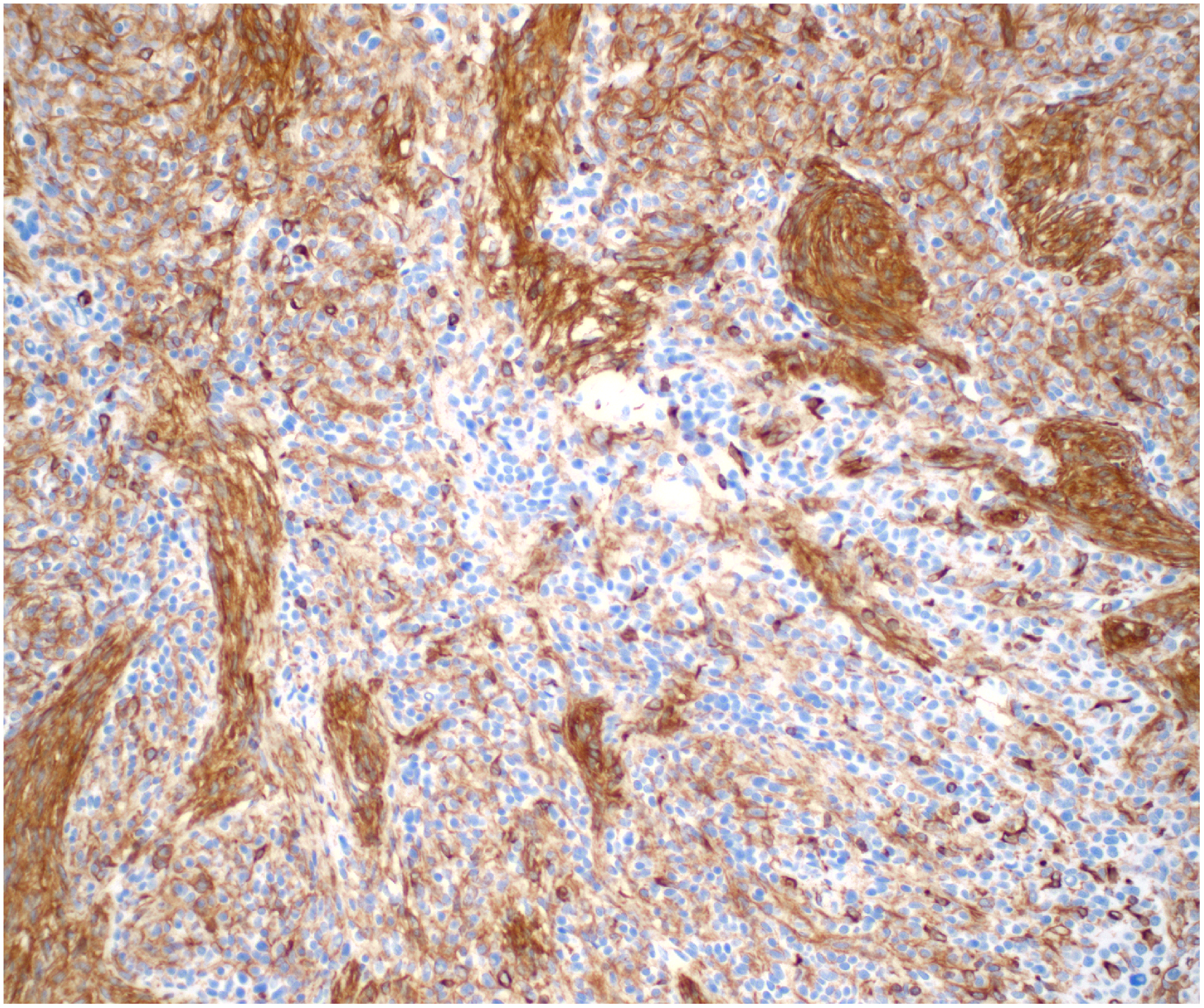
Vimentin stain, 20×. In the metaplastic process of epithelial-mesenchymal transition (EMT), transdifferentiated spindle cells up-regulate expression of mesenchymal markers including vimentin.
Vivero et al were the first to describe a recurring fusion between YAP (Yes Associated Protein-1) and MAML2 (Mastermind Like Transcriptional Coactivator 2) genes in 2020. 13 Among 8 patients with metaplastic thymoma, fusions were detected by DNA sequencing in 4 cases. In 2 cases, there was no fusion or somatic mutations detected on DNA sequencing, however they identified RNA transcripts fusing exon 5 of the AP1 gene and exon 2 of the MAML2 gene. In the remaining 2 cases, no fusion was detected. They compared this genomic profile to those of 52 thymic neoplasms (25 thymomas, 25 thymic carcinomas, and 2 thymic neuroendocrine carcinomas). They found TP53 mutations in 9 cases (36%) and CDNK2A deletion in 7 cases (28%), but no fusion events. A second study conducted by Zhao et al of 17 patients in 2021 identified YAP1::MAML2 translocation in 13 out of 17 patients with a diagnosis of metaplastic thymoma, but in zero of 9 patients of type A thymomas, and zero of 7 cases of micronodular thymoma with lymphoid stroma. 16 Using FISH method in microdissection slides, they showed that both epithelioid and spindle cell components harbored YAP1::MAML2 fusions. These findings further support classification of metaplastic thymoma separately from other thymomas.
The differential diagnosis for metaplastic thymoma should include thymic tumors (thymoma type A, type AB, thymic carcinoma, thymic carcinosarcoma, or sarcomatoid carcinoma) as well as mediastinal tumors such as biphasic mesothelioma, synovial sarcoma, teratoma. The lack of significant atypia, high mitotic activity, foci of necrosis, and use of appropriate immunostains can help distinguish metaplastic thymoma from other malignant processes. Thymoma type A or type AB may be more difficult to rule out, particularly if the metaplastic thymoma has a prominent spindle cell component. Extensive sampling may be of help to appreciate the characteristic biphasic pattern on metaplastic thymoma. Due to the presence of a gene fusion YAP1::MAML2, molecular studies further assist in diagnosis.
Myasthenia gravis occurs in association with thymoma in upwards of two-thirds of patients, but is rarely found in metaplastic thymoma making our patient a rarity. Only 3 cases prior to the present patient have been described with myasthenia gravis, and all of those were women aged 44 or older. Including the present case, 3 have had serum acetylcholine receptor antibodies. Lymphocyte-poor and lymphocyte-rich thymoma with associated myasthenia gravis are correlated with the activation of different functional pathways with immunological relevance. 17 In type A thymomas without active thymopoeisis, a link has been proposed between myasthenia gravis and the population of AChR-reactive peripheral T lymphocytes present in most people. Upon recirculation to the thymoma, these lymphocytes may become activated and lead to myasthenia gravis. 18 Additionally, thymopoiesis in type A thymomas can be focal, indicating that evidence of the process may be missed without complete sampling. The autoimmune regulator protein (AIRE) is absent in essentially all thymomas whether or not a patient has myasthenia gravis. AIRE is normally involved in the intrathymic selection of regulatory T cells. Thymoma-associated myasthenia gravis depends on the intratumorous generation and subsequent export of CD4 + effector T cells, which are directed against acetylcholine receptor epitopes from the thymoma to the peripheral immune system. The deficiency of regulatory T cells would be expected to promote a breakdown in tolerance and development of autoantibodies and the autoimmune disease.
In metaplastic thymoma, intratumoral immature T-lymphocytes are usually not present or scarce. 4 Another report of metaplastic thymoma described the presence of TDT-positive lymphoid cells, which the authors suggested may have driven the presence of myasthenia gravis in their patient. 12 After extensive microscopic examination of our patient's thymoma, few TDT-positive lymphoid cells were observed in the periphery of the tumor, consistent with remnants of thymus parenchyma uninvolved by tumor. It is likely that the high doses and prolonged treatment with prednisone could have led to a reduction of lymphocytes in this patient. We cannot exclude that more extensive sampling could have led to detection of these cells.
Conclusion
The patient has several atypical features. He is relatively young compared to the other reports and is a male. There was a lack of TDT-positive T lymphocytes and lymphocytic infiltrate generally, which are features of autoimmune disease associated with thymoma. These cells are the presumed drivers of intratumorous thymopoiesis with a failure of tolerance development as the expected etiology. He also has associated myasthenia gravis. While autoimmune disorders are commonly associated with thymoma, they remain a rarity among metaplastic thymoma patients. These unusual features present opportunities for further exploration of the link between metaplastic thymoma and myasthenia gravis.
Footnotes
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Henry J. Kaminski, M.D., is a consultant for Roche, Cabeletta Bio, and UCB Pharmaceuticals; and is CEO and CMO of ARC Biotechnology, LLC based on US Patent 8,961,98. He is principal investigator of the Rare Disease Network for Myasthenia Gravis (MGNet) National Institute of Neurological Disorders & Stroke, U54 NS115054, Targeted Therapy for Myasthenia Gravis. R41 NS110331 to ARC Biotechnology, and co-investigator for R43NS124329 MV2C2 antibody as a new therapeutic for myasthenia gravis to Mimivax, LLC. Keith Mortman, M.D., is a consultant for Ethicon & on the advisory board for Guardant Health. We have no other potential conflicts to disclose.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the MGNet, a member of the Rare Disease Clinical Research Network Consortium (RDCRN) NIH U54 NS115054.
Ethical Approval
Not applicable, because this article does not contain any studies with human or animal subjects.
Informed Consent
Not applicable, because this article does not contain any studies with human or animal subjects.
Trial Registration
Not applicable, because this article does not contain any clinical trials.