
Abstract
Spermatocytic tumour (ST) accounts for 1% of testicular germ cell tumours. It is an indolent neoplasm with good prognosis. In approximately 6% of STs, sarcomatous dedifferentiation may occur, portending an aggressive behaviour and representing a significant diagnostic challenge that can lead to its misdiagnosis. Herein, we report the clinicopathological features of a patient with a sarcomatoid spermatocytic tumor, initially diagnosed as mixed germ cell tumour, who was referred to our institution with lung metastases mainly composed of rhabdomyosarcomatous elements. This case report illustrates the importance of recognizing this entity for adequate management of these patients.
Keywords
Introduction and Epidemiology
Testicular tumours are rare neoplasms, accounting for only 1% of all tumours in men. 1 Most are classified as germ cell tumours. The World Health Organization (WHO) subclassifies germ cell tumours as derived from germ cell neoplasia in situ or not. The latter include spermatocytic tumour (previously known as spermatocytic seminoma). 2
Spermatocytic tumour, although previously associated with classical seminoma, is now confirmed to be a clinically and pathologically distinct entity,3,4 with no relationship either with seminoma or other germ cell tumours derived from germ cell neoplasia in situ (GCNIS-derived). 2
Spermatocytic tumour accounts for 1-2% off all germ cell tumours. 5 Typically, it occurs in the fifth or sixth decades of life and has an indolent course and good prognosis.5,6 In approximately 6% of STs, a sarcomatous dedifferentiation develops, increasing the risk for metastasis and worsening the prognosis. 5
We report the case of a patient with a sarcomatoid spermatocytic tumour, presenting with a large testicular mass and multiple pulmonary metastasis, initially misdiagnosed as mixed germ cell tumour. As so, we highlight the importance of a correct diagnosis for patient treatment and follow-up.
Case Report
A 46-year-old man presented with a slowly growing painless and enlarged right testis mass. There was no relevant personal or family medical history.
Tumour markers α-fetoprotein (AFP) and beta-human chorionic gonadotropin (β-HCG) were normal, and lactate dehydrogenase (LHD) was two times above the upper limit of normal.
A scrotal ultrasound revealed an 18 × 13 × 8 cm heterogenous multinodular mass, with cystic areas. Differential diagnosis between primary testicular cancer and lymphoma of the testis was suggested and the patient underwent orchiectomy.
Initial histologic diagnosis was of a mixed germ cell tumour with areas of seminoma, yolk-sac tumour and teratoma. Afterwards, the patient was referred to our institution, for further treatment and follow-up.
For stagging purposes, a thoraco-abdominopelvic CT was performed and revealed multiple bilateral pulmonary masses up to 3 cm in size, as well as enlarged mediastinal lymph nodes.
In our institution, slides from the orchiectomy were reviewed and exhibited a distinctive spermatocytic tumour composed of solid and pseudo-glandular areas with large cells, intermediate cells and small lymphocyte-like cells, positive for SALL4 and KIT and negative for PLAP and CD30 (Figure 1). No germ cell neoplasia in situ was identified. Additionally, a high-grade sarcoma component was observed, comprised of pleomorphic and spindle cells, some also with rhabdoid features, focally positive for DES, MYOD1, MYOG and SALL4 (Figure 2). A final diagnosis of spermatocytic tumour with rhabdomyosarcomatous differentiation was rendered. Simultaneously, a lung biopsy was performed, revealing a germ cell tumour metastasis composed almost exclusively of a rhabdomyoblastic component with strong and diffuse positivity for DES, MYOG and MYOD1 and focal SALL4 expression (Figure 3).
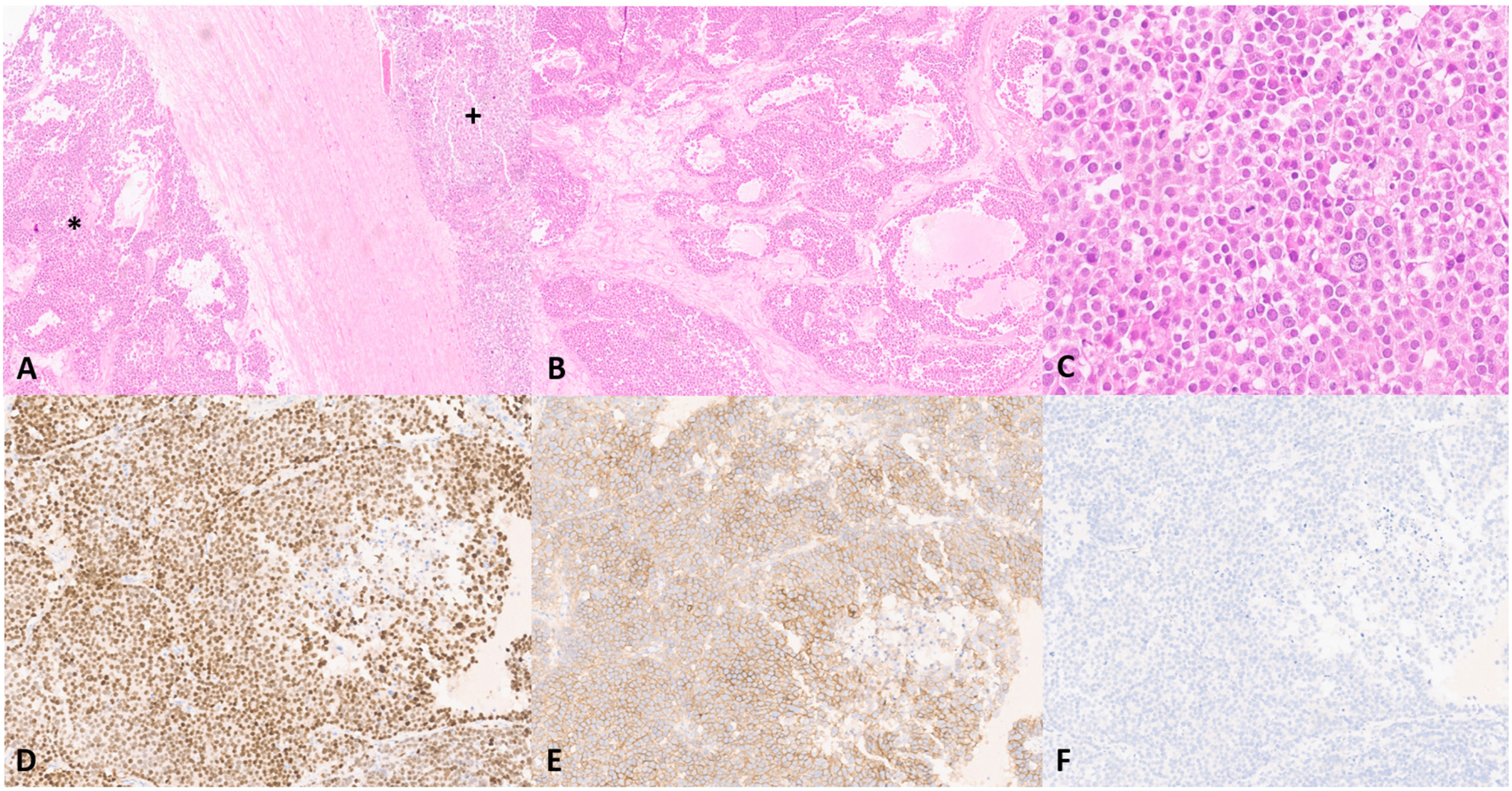
Testicular sarcomatoid spermatocytic tumor. Two juxtaposed areas of spermatocytic tumor on the left side (*) and sarcomatoid component on the right ( + ) (A). Classic spermatocytic tumor shows a diffuse to multinodular growth pattern, with pseudoglandular areas and stromal oedema (B). Higher magnification shows a polymorphous cell population, consisting of three distinct cell types based on cell size and chromatin (C). SALL4 is strongly and diffusely positive in spermatocytic tumor (D) as well as KIT (E). Keratin is negative (F).
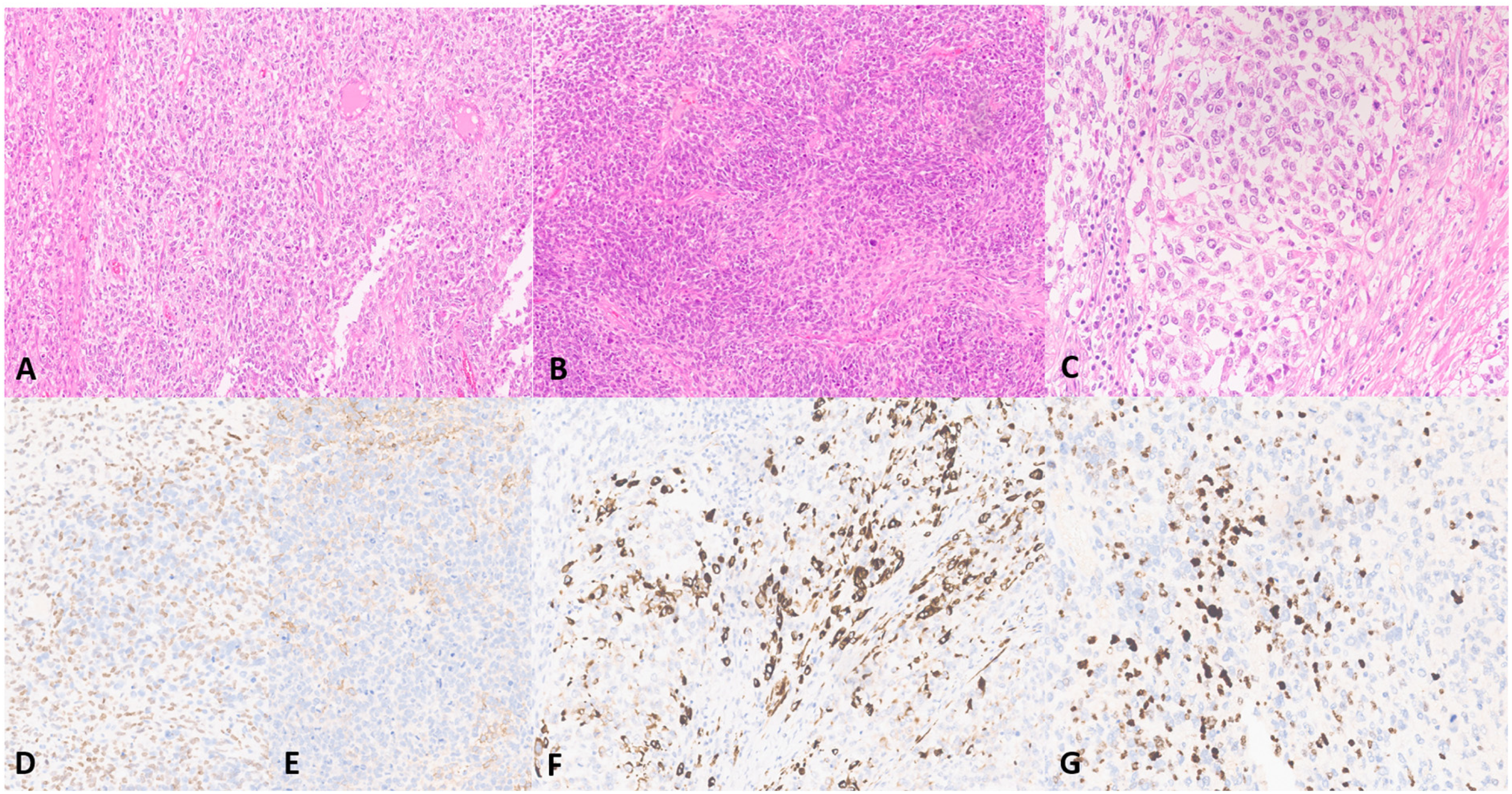
Testicular sarcomatoid spermatocytic tumor. Solid and diffuse areas of epithelioid and pleomorphic cells (A), along with fascicular areas with spindle cells (B) and focal rhabdoid morphology (C). SALL4 (D) and KIT (E) were focal and weakly positive in sarcomatoid components. Focal expression of DES (F), MYOD1 (G) and MYOG was shown in areas with rhabdoid features.
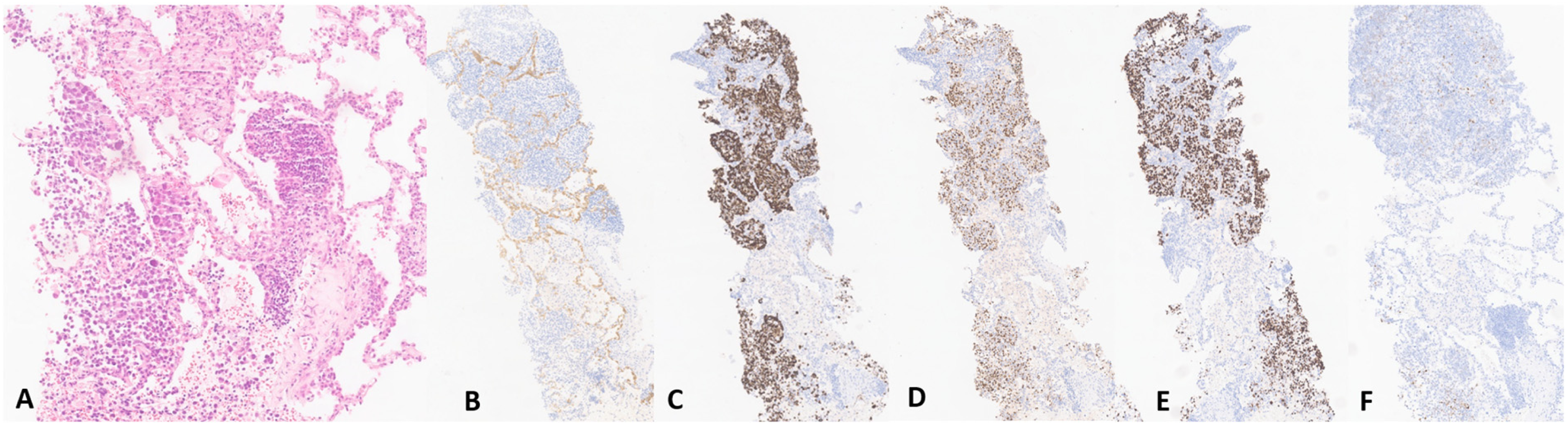
Pulmonary metastatic germ cell tumor with rhabdomyoblastic differentiation. A epithelioid to rhabdoid neoplasm is seen infiltrating alveolar spaces (A). The immunohistochemical staining shows negativity for keratin AE1/AE3 (B) and positivity for DES (C), MYOG (D) and MYOD1 (E). Focal positivity was seen for SALL4 (F).
The patient underwent 4 cycles of chemotherapy with VIP (etoposide + cisplatin + ifosfamide), showing favourable response of the lung lesions, but no response in lymph node metastases. Three months after treatment, he presented with neurological symptoms and central nervous system metastases were identified. Whole-brain radiation therapy was begun, but the patient died due to infectious and respiratory complications.
Discussion
Spermatocytic tumour was initially described in 1946 by Masson 7 and accounts for 1-2% off all germ cell tumours. 5 Unlike other germ cell tumours, it typically occurs later in life, as it did in our patient, although it has been reported in a wide range of ages, from 19 to 92 years-old. 6
These neoplasms are thought to derive from more differentiated germ cells, such as spermatocytes and spermatogonia.5,8,9 Genetically, amplification of the DMRT1 gene on chromosome 9p is common in spermatocytic tumours but chromosome 12p amplification, typical seen in GCNIS-derived tumours, is not present. However, positivity has been reported for spermatocytic tumours with anaplastic transformation.10,11 It is hypothesised that mutations in age-related genes, such as FGFR3 and HRAS, may promote tumour development. 12
It is usually an indolent tumour. 9 Patients often present with a slowly growing testicular mass.9,13 It does not affect pre-pubertal patients and cryptorchidism is not a risk factor. 14 Also, it has no ovarian counterpart, unlike other tumours, such as seminoma.4,9 It is typically unilateral with a slight tendency to occur in the right testis, despite reported cases of bilaterality, usually metachronous. 15 Tumour markers, as β-HCG and AFP are typically normal,9,13 although some case reports have found elevated LDH and/or AFP. 16 Our patient also presented with a right testis mass and no tumour marker elevations were documented.
Grossly, it presents as a testicular mass with a size ranging from 1.5 to 28 cm (average 7 cm). 9 It is usually well circumscribed and confined to the testis. Tumours are often tan-grey, soft, friable, and multilobulated, with gelatinous regions. 17 Rarely, especially with larger masses, necrotic and haemorrhagic foci may be found. 9
Microscopically, spermatocytic tumour has a heterogeneous population composed of three types of cells: small (6-8 µm), intermediate (15-18 µm) and large (50-100 µm). 9 Neoplastic cells are arranged in sheets with areas of oedema in the background. 17 Unlike classic seminoma, there are scarce or absent lymphocytes, no granulomas and no fibrovascular septa.16,18 Two types of tumour infiltration have been described, namely interstitial and intratubular, the latter being more common, and must be distinguished from germ cell neoplasia in situ, which is not associated with spermatocytic tumour.9,17
Immunohistochemically, spermatocytic tumour shows negativity for OCT4, CD30, smooth muscle actin, DES, β-HCG, VIM, human placental lactogen, leucocyte common antigen, keratin AE1/AE3, NSE, ALPP, and AFP.5,9,15,17 SALL4 is positive. 17 KIT positivity has been reported.9,15
Standard treatment for spermatocytic tumours is orchiectomy followed by surveillance. 4 In case of metastatic disease, radical orchiectomy followed by adjuvant chemotherapy may be beneficial. 19 These tumours have good prognosis due to the high effectiveness of orchiectomy and their low propensity to metastasize.4,5,8
Sarcomatous Dedifferentiation
Sarcomatous dedifferentiation is a rare event that confers tumour aggressiveness, often presenting with metastatic disease. 5 Its pathogenesis is still unknown, although there are some hypotheses. The most widely accepted states that sarcomatous component may arise from anaplastic transformation or dedifferentiation of spermatocytic tumour cells.5,16,18–21 Currently, no genetic alterations have been described.
To date, most of the literature regarding sarcomatous spermatocytic tumour is based in case reports or small series. At least 24 reported cases have been described and we believe our patient is the 25th (Table 1). In these cases, spermatocytic tumour occurred in patients with a mean age of 52 years and metastatic disease was frequently documented in lung, lymph nodes, liver and bone. Most dedifferentiated components were undifferentiated spindle cell and rhabdoid neoplasms. Out of the 25 patients, 9 were alive after a mean follow-up of 11 months and 11 patients died of the disease after a mean follow-up of 9 months. This data was not available for the remaining 5 patients. Our patient showed a tumour with spindle/rhabdoid features, lung and lymph node metastases, and is only the second case with reported brain metastases.
Cases Summary of ST with Sarcomatous Dedifferentiation.
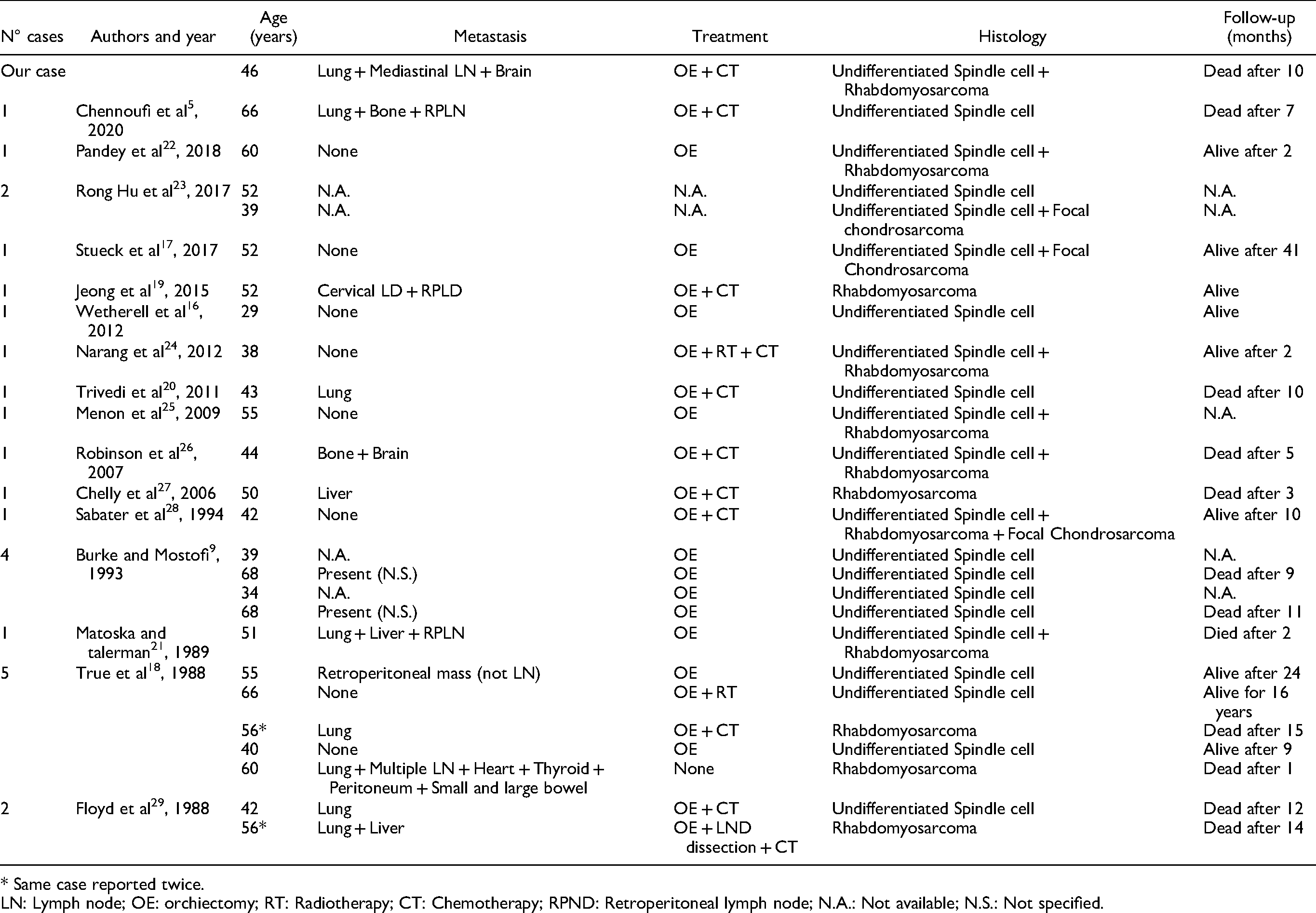
* Same case reported twice.
LN: Lymph node; OE: orchiectomy; RT: Radiotherapy; CT: Chemotherapy; RPND: Retroperitoneal lymph node; N.A.: Not available; N.S.: Not specified.
The differential diagnosis includes other tumours with sarcomatous appearance, like a malignant somatic component of teratoma or primary sarcomas of the testis and paratestis. 17
Standard treatment for localized sarcomatous spermatocytic tumours is radical orchiectomy. However, since there is higher probability for metastasis, chemotherapy is usually considered. Although no specific chemotherapy combination appears to be particularly effective, an association between etoposide, ifosfamide and cisplatin (VIP) appears to present the best results. 19 Our patient was treated with orchiectomy followed by chemotherapy in a similar scheme, initially showing favourable response in lung metastases. Nevertheless, despite aggressive therapy, our patient died 10 months after diagnosis.
Conclusion
Spermatocytic tumours are rare neoplasms. Even rarer is the sarcomatous dedifferentiation, conferring a higher rate of metastatic disease and worse prognosis. 5 Although clinically sarcomatous spermatocytic tumours may not show distinctive features, some characteristics as rapid growth or aggressive metastatic disease may raise a suspicion for this entity. 5 Histological awareness and recognition of this tumour with the exclusion of important differential diagnosis is fundamental for adequate treatment and follow-up.
Our case highlights the importance of histological diagnosis of sarcomatous spermatocytic tumours, particularly in older patients. As most of the literature is still based on case reports, further studies are needed in order to improve our knowledge of the most adequate management of patients with spermatocytic tumours with sarcomatous dedifferentiation.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.