
Abstract
Objective
To develop consensus-based algorithms for genetic testing in patients with common craniofacial conditions.
Design
An online collaborative consisting of online meetings, independent work, and feedback across groups. Setting/Participants: A collaborative of genetics and pediatrics providers from three regional craniofacial centers (four institutions).
Methods
Collaborative participants agreed upon a shared initial framework, developed algorithms independently, and presented/tested the algorithms with a national audience. Algorithms were modified based on consensus feedback.
Results
The collaborative group developed final algorithms for genetic testing in patients with orofacial cleft, branchial arch conditions, and craniosynostosis.
Conclusions
Timely and accurate diagnosis of genetic conditions can support medical management recommendations that result in safer surgical interventions. Algorithms can help guide best-practices for testing, particularly in institutions without easy access to genetics providers.
Objective
The majority of patients cared for by multidisciplinary craniofacial teams have conditions involving orofacial clefting, craniosynostosis, or branchial arch anomalies. For each patient, team providers must determine if symptoms represent an isolated condition or are signs of an underlying genetic syndrome. Determining the need for and timing of genetics consultation is often complex but can play an important role in surgical planning and medical management. For example, surgical management of a child with cleft palate associated with chromosome 22q11.2 deletion syndrome would include cardiovascular and spine imaging studies in addition to screening for hypocalcemia and immunodeficiency. Recognition of children with craniofacial conditions at risk for genetic syndromes and associated health concerns that could impact surgical safety is critical and can be time-sensitive. The objective of our study is to provide a framework for providers regarding indications for genetic assessment in individuals with craniofacial conditions using condition-specific algorithms. Clinical algorithms (flow charts) are important tools that assist healthcare providers in the evaluation of a patient's clinical health concern(s). Examples of clinical algorithms for pediatric patients that contain genetic testing recommendations include: “Macrocephaly in infants and children: Etiology and evaluation” 1 and “Intellectual disability in children: Evaluation for a cause” 2 currently available through the online resource, UpToDate, and- a clinical algorithm for the genetic workup for congenital heart disease proposed Jerves. 3 In this project we sought to design genetic testing algorithms for pediatric patients with orofacial clefting, craniosynostosis, and branchial arch anomalies. The design of each algorithm was focused on determination of an accurate diagnosis, including when to recommend genetic testing, to allow early identification of medically actionable recommendations that could impact surgical planning and improve patient outcomes, and potentially provide more accurate assessment of recurrence risk. For each algorithm, we will share examples of the potential impact of genetic diagnoses on medical and surgical management for patients seen in craniofacial clinics and we note importance of future validation studies and assessment of impact on patient outcome. 4
Design
Specialists in craniofacial medicine from three regional centers designed genetic testing algorithms for children with craniofacial conditions that fit within three major categories: craniosynostosis, orofacial clefting, and branchial arch anomalies. Each regional working group included a pediatrician, clinical geneticist, and genetic counselor. The regional working groups were assigned the task of designing one of three testing algorithms. One algorithm was designed for children presenting with craniosynostosis. A second algorithm was designed for children with cleft lip and/or cleft palate, and the third algorithm was designed for children with an anomaly of the branchial arch (ear, neck, lower face and jaw). Algorithms were designed based on review of current literature and clinical expertise. Over the course of six months, each algorithm was presented to the entire panel of specialists for extensive review and revision. Modified algorithms were pilot tested at each institution over a 6-month period and secondarily revised. Revised algorithms were presented in a workshop at the American Cleft Palate-Craniofacial Association (ACPA) 79th Annual Meeting in 2022 to gain feedback from providers. Workshop participants had the opportunity to apply the algorithms in the assessment of provided case scenarios and provide feedback to further revise algorithms.
Setting
Craniofacial providers from four institutions (3 regional centers) participated in this study. The institutions involved were Seattle Children's Hospital, University of California Los Angeles, Children's Hospital Los Angeles, and University of California San Francisco Benioff Children's Hospitals. Each of these institutions houses a multidisciplinary craniofacial clinic that provides care to pediatric patients.
Patients/Participants
After the three algorithms were designed, each was presented to providers from the 3 regional centers and underwent first revision based on feedback. Each institution then piloted each of the three algorithms with patients seen in their respective clinics. Each algorithm was applied to two groups of patients: (1) those with an established genetic diagnosis (N = 35) and (2) those presenting for initial assessment without established diagnosis (N = 66).
This work was approved by the Institutional Review Board (IRB) at the University of California San Francisco. The IRB at Seattle Children's Hospital and the University of California Los Angeles determined this to be quality improvement work not requiring review. The IRB at the Children's Hospital of Los Angeles declined to review this retroactively.
Interventions and Main Outcome Measures
Genetic Testing Algorithm Development Process
Three working groups (each comprised of pediatrician, clinical geneticist, and genetic counselor) were assigned the task of designing a testing algorithm for one of the following conditions (orofacial clefting, branchial arch conditions, or craniosynostosis). The initial algorithm design process included: (1) review of current literature, (2) review of frequency of underlying genetic causes, (3) exploration of potential impact of genetic testing on patient care, and (4) optimal genetic test for each condition to achieve diagnosis. Each proposed algorithm (version 1, v1) was presented to the entire working group for extensive discussion, revision, and review over a 6-month period. Revised algorithms (version 2, v2) were shared between centers and pilot tested over the 2nd 6-month interval. Pilot testing included the application of the algorithms in two groups of patients: (1) those with an established genetic diagnosis (N = 35; 11 with craniosynostosis; 15 with orofacial clefting; 9 with branchial arch anomalies) and (2) those presenting for an assessment without established diagnosis (N = 66; 27 with craniosynostosis; 27 with orofacial clefting; 12 with branchial arch anomalies). Two questions were assessed for Group 1 patients (1) did the algorithm lead to the correct diagnosis, and (2) what number of genetic testing steps were needed to reach diagnosis. For Group 2 patients, providers were asked if they agreed with the testing recommended by the algorithm. A total of 101 patients were reviewed (1/3 with established diagnoses) and algorithms were revised (version 3, v3) to maximize identification of genetic diagnosis and minimize number of steps to diagnosis. The v3 algorithms were presented during a 90-min study session at the 2022 ACPA conference. Attendees had the opportunity to test algorithm with sample cases and make suggestions for improvement.

Orofacial Clefting Genetic Testing Algorithm
Cleft lip and/or cleft palate results from failure of complete fusion of the maxillary first brachial arch with the frontonasal process in early gestation (Figure 1). 5 Cleft lip and/or cleft palate can be non-syndromic, or syndromic when they are associated with additional anomalies and/or neurodevelopmental disorders. Cleft palate only is more likely to be associated with a syndrome, compared to cleft lip with or without palate.6,7 Syndromic forms of orofacial clefts are more likely to be associated with chromosomal abnormality or coding variant in a single gene and therefore may complicate an individual's medical or surgical needs.6–8
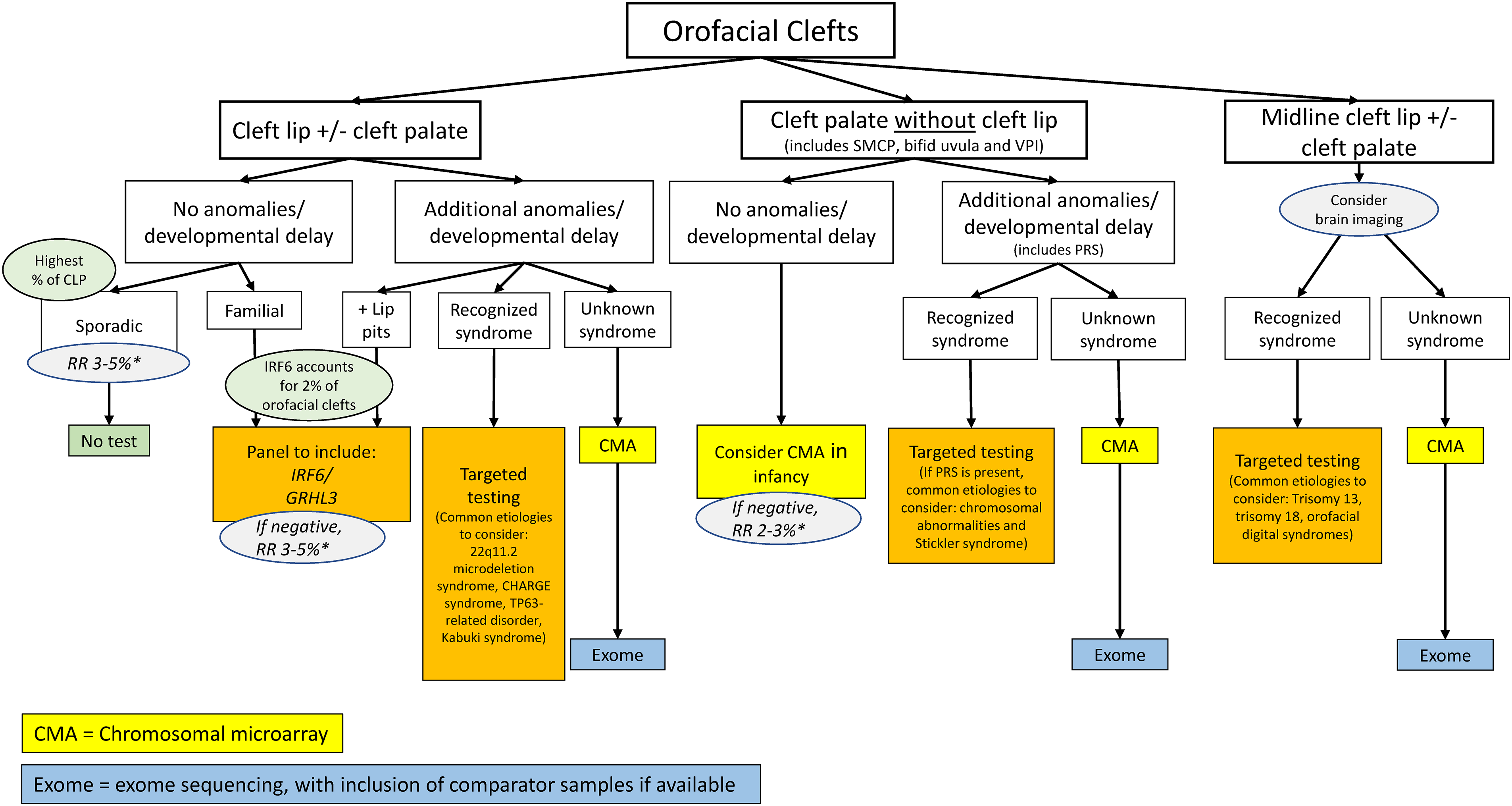
Orofacial cleft genetic testing algorithm. This algorithm should be used for individuals with cleft lip with or without cleft palate, cleft palate, and midline cleft lip with or without cleft palate. Individuals who have palatal abnormalities including submucous cleft palate (SMCP), bifid uvula and velopharyngeal insufficiency (VPI) are considered under cleft palate group in this algorithm. Pierre Robin sequence (PRS) is associated with micrognathia, cleft palate, and glossoptosis that may lead to respiratory obstruction. In this algorithm, PRS is considered as additional anomaly to cleft palate when present. *Empiric recurrence risk.
The
Branchial Arch Genetic Testing Algorithm
The branchial arches are the embryological precursors of the face, neck, and pharynx. Derivatives of the first arch include the maxillary and mandibular processes of the jaw, muscles of mastication, malleus, incus, external auditory canal, facial bones and cartilage, and cranial nerve V. Derivatives of the second arch include the muscles of facial expression, hyoid arch, tonsillar fossa, stapes, and cranial nerves VII and VIII (Figure 2).9,10 Major craniofacial anomalies involving the first and second arch structures include hypoplasia of the mandible (micrognathia), failure of maxillary fusion, and auricular atresia. Examples of common first and second arch syndromes include Craniofacial microsomia, Treacher Collins syndrome, Branchio-oto-renal syndrome, EFTUD2-related disorders, and Townes-Brocks syndrome.11–16
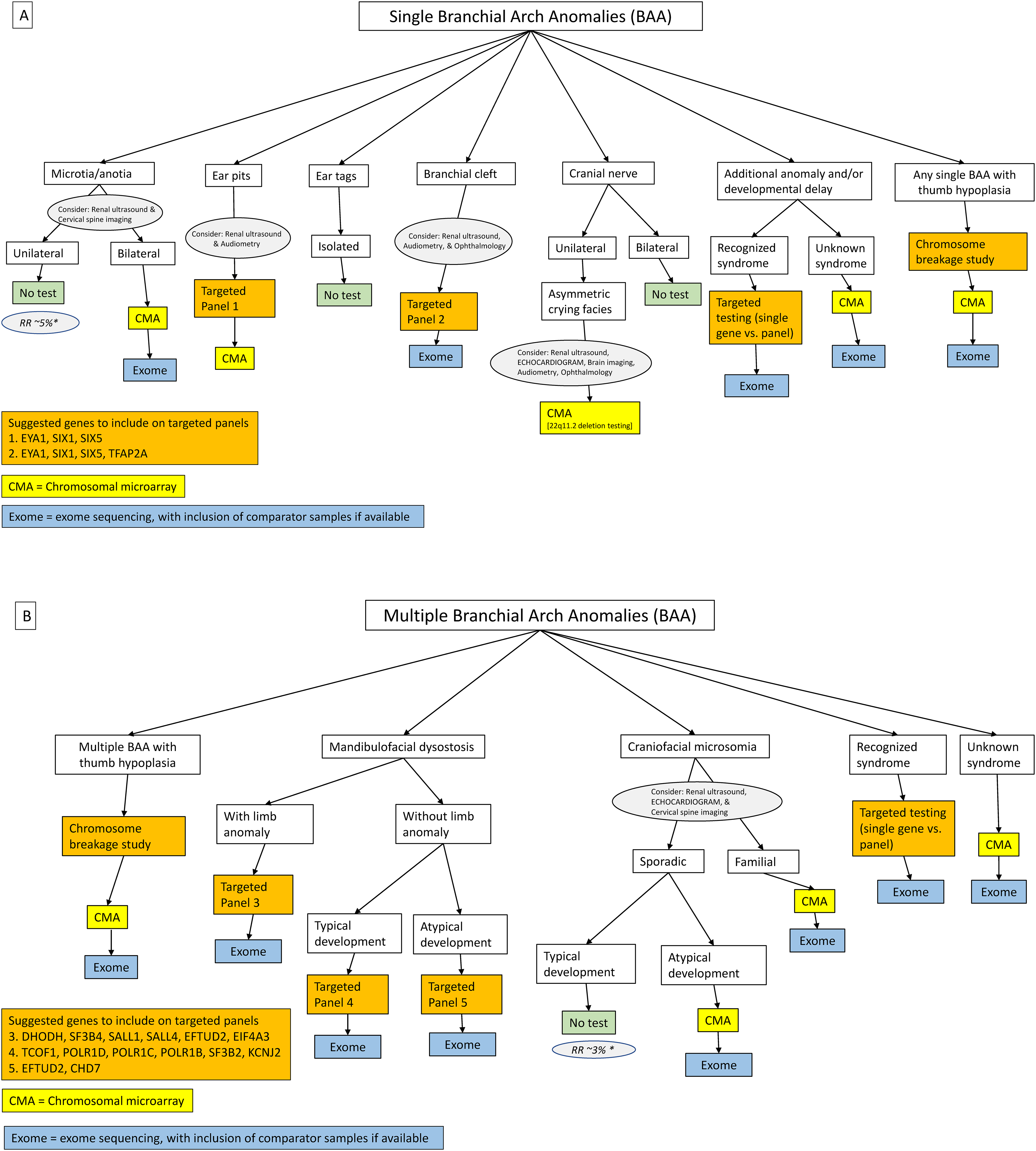
Branchial arch anomaly genetic testing algorithms. This algorithm should be used for individuals with single branchial arch anomaly (BAA). For individuals with specific single BAA (microtia, ear pits, branchial cleft, and unilateral facial palsy), recommendations for targeted imaging studies and/or subspecialty consultation is indicated in circles. *Empiric recurrence risk B. This algorithm should be used for individuals with multiple branchial arch anomalies. For individuals with clinical diagnosis of Craniofacial Microsomia recommendations for targeted imaging studies are indicated in circle. *Empiric recurrence risk.
The
Pilot testing of
Craniosynostosis Genetic Testing Algorithm
Craniosynostosis (CS) refers to the premature closure of a single or multiple cranial sutures (Figure 3). 26 Sagittal, coronal, metopic, and lambdoid (from most to least common, respectively) sutures may be involved.27–30 Multisuture CS occurs when two or more sutures are involved and includes bicoronal CS. Craniosynostosis is non-syndromic (NSCS) in 70% of cases26,27,31 and syndromic (SCS) in 30% of cases. Twenty-five percent of individuals with CS have an identifiable genetic cause.26–28
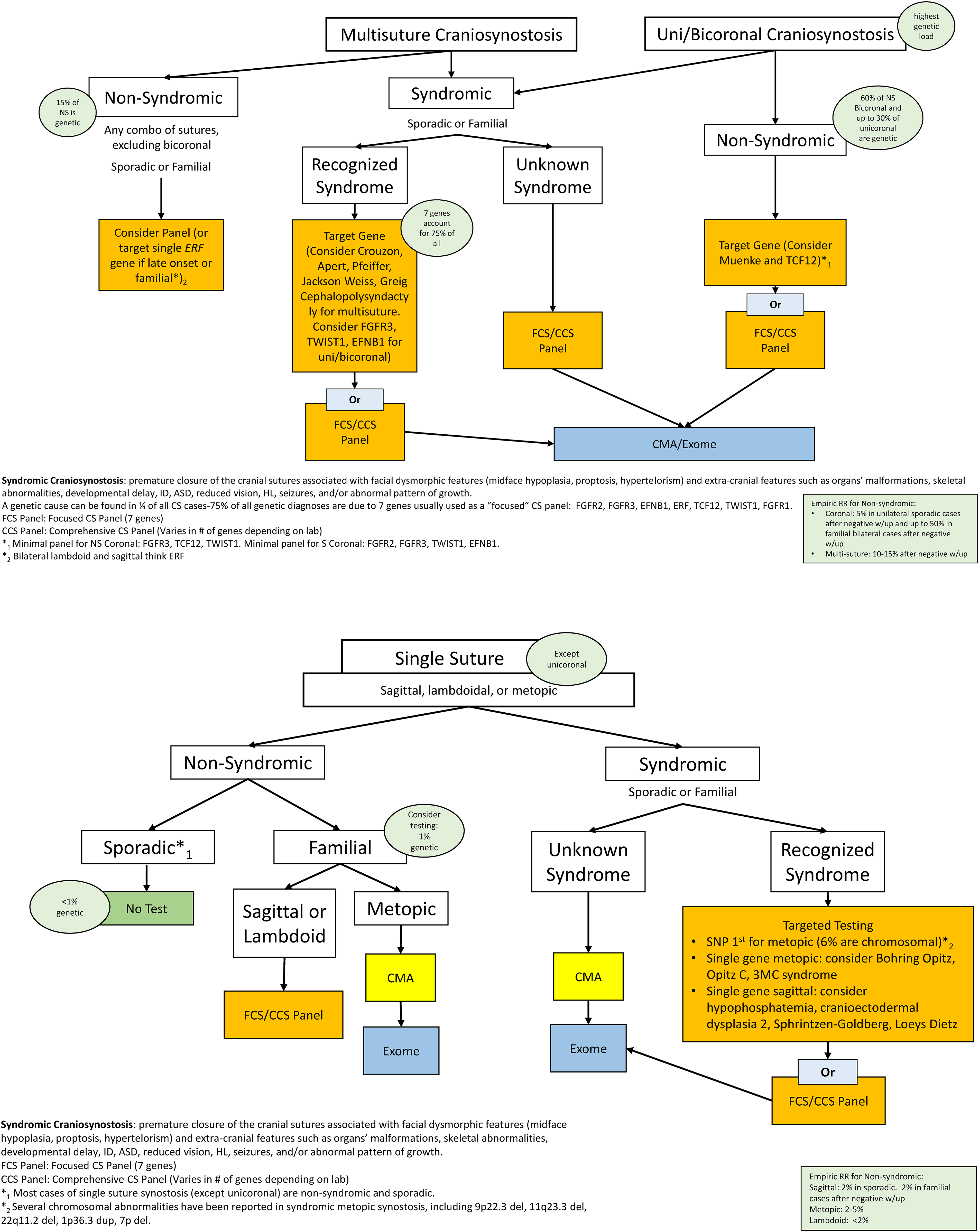
Craniosynostosis genetic testing algorithms: the multisuture craniosynostosis algorithm should be used for individuals with more than one suture involved as well as in individuals presenting with unilateral coronal synostosis which is treated the same way than bilateral coronal synostosis given the similar genetic load. The single suture algorithm should be used for individuals with a specific single suture synostosis, except for coronal synostosis.
Of those individuals initially classified as NSCS, 10-15% are subsequently found to have a pathogenic variant especially if NS bicoronal, unicoronal, and multisuture CS is involved.27,28 SCS may be associated with dysmorphic facial features (eg, midface hypoplasia, proptosis, hypertelorism) and extra-cranial features (eg, organ malformations, skeletal abnormalities, seizures, hearing loss, developmental delay, intellectual disability, autism spectrum disorder, reduced vision, hearing loss, seizures, and/or abnormal growth pattern).26–30,32–34
The initial
Discussion
Although many children with craniofacial conditions receive multidisciplinary team care, access to clinical geneticist may vary and frontline team providers need to determine if the child's findings represent an isolated or syndromic condition with potential for additional health concerns. For each child, the provider must make recommendations for assessments ranging from laboratory tests, imaging, and subspecialty consultation to assure that the child is safe and ready for surgical procedures and guide medical management. This includes need for and timing of genetics consultation and testing. Access to a flow chart, or algorithm, to guide genetics consultation and testing could help to identify high risk children. We recommend consultation of the algorithm and consideration of testing as soon as a condition is identified, and that the algorithm be revisited if new findings are identified.
When a team provider evaluates a child with orofacial clefting, they must determine if a child is at risk for additional health concerns promptly as cleft lip and palate surgery occurs within the first year of life. For example, an infant born with cleft palate and congenital heart anomaly should have testing for chromosome deletion/duplication syndromes including 22q11.2 microdeletion syndrome. Infants with 22q11.2 deletion syndrome have significant risks for associated health concerns including immunodeficiency, hypocalcemia, cervical spine instability that would impact medical and surgical management. 36 If a patient with a cleft palate has facial asymmetry, dysplastic ears, and hypotonia, genetic assessment could lead to diagnosis of CHARGE syndrome with concerns for dysphagia and risk for aspiration events, congenital heart disease, vision and hearing impairment, and immunodeficiency. Different health concerns would be considered for a child presenting with a midline cleft lip. This child could also have midline brain defects such as holoprosencephaly with risk for hypopituitarism 37 or be at risk for renal insufficiency if the diagnosis is oro-facial-digital syndrome.8,38 Thus, the presence of a midline cleft lip should lead to brain imaging and, if the child has holoprosencephaly, testing for hypopituitarism must be pursued prior to surgery dueto the risk for life-threatening central diabetes insipidus, cortisol deficiency, or hypoglycemia due to growth hormone deficiency. For each of these orofacial clefting conditions, early testing and diagnosis would result in condition-specific recommendations that would improve surgical safety and guide management.
Although unilateral microtia can be an isolated finding, when an infant with unilateral microtia has additional health concerns, genetic assessment should be considered. For example, an infant presents to team for evaluation of unilateral microtia and is found to have small thumbs and history of horseshoe kidney on prenatal ultrasound. Given these findings, genetic consultation is pursued with subsequent recommendation for chromosome breakage studies due to concerns for Fanconi anemia. Children with Fanconi anemia are at high risk for bone marrow failure and leukemia and urgent referral to hematology-oncology service is recommended. Additionally, many of the branchial arch conditions present with overlapping features and genetic consultation and testing is important to achieve correct diagnosis to guide medical management. Consider a scenario in which a child with bilateral microtia, cleft palate, mild maxillary hypoplasia, and mandibular hypoplasia presents for team assessment. Although initial clinical suspicion is Treacher Collins syndrome, review of growth chart shows microcephaly and subsequent genetic testing reveals a pathogenic variant in the EFTUD2 gene. Infants with EFTUD2-related disorder are at an increased risk of tracheoesophageal fistulae, developmental delays, and cervical spine anomalies whereas infants with Treacher Collins syndrome are not. Thus, a specific genetic diagnosis for this child provides guidance for screening studies important in planning safe surgeries and supporting health and development.
Syndromic CS syndromes may not be difficult to suspect clinically but considerable phenotypic overlap exists,26–29,32 therefore confirmation of specific gene variants is important for disease prognosis and management.26–28,34 For example, Pfeiffer syndrome associated with the W290C variant in the FGFR2 gene is associated with high risk for tracheal cartilaginous sleeve, which confers significant mortality risk that can be mitigated with the appropriate intervention.26,39–41 Crouzon syndrome confirmed molecularly is important in atypical cases due to the increased risk of Chiari I malformation and progressive hydrocephalus (30%) often with tonsillar herniation in many FGFR2 variants. 32 Crouzon syndrome with Acanthosis Nigricans due to the A391E variant in FGFR3 is associated with higher risk of Chiari 1 malformation, hydrocephalus, choanal stenosis/atresia, and increased risk of odontogenic tumors later in life.32,42–44
Two conditions that may present with variable expressivity and a mild phenotype and/or nonsyndromic coronal CS are: 1) Muenke syndrome and 2) TCF12. 27 Muenke syndrome is the most common syndromic form of SCS caused by a single amino acid substitution (P250R variant in FGFR3) and in 65% is familial and can present with only macrocephaly without craniosynostosis in some affected individuals. Greater than 70% of individuals with Muenke syndrome have hearing loss (majority bilateral sensorineural HL (SNHL) and some progressive). 27 A confirmed molecular diagnosis allows for accurate recurrence risk, assessing for risk of SNHL, developmental delays, ADHD, and seizures.30,32,45–48 TCF12 has variable penetrance, with non-penetrance in 50% of cases and absent craniosynostosis in some. A confirmed diagnosis of TCF12 will inform about the risk of learning disabilities, autism spectrum disorder, Chiari 1 malformation, intracranial pressure with risk of visual sequelae, and developmental delays.27,49,50
There should be a high risk of suspicion for underlying chromosome imbalance in infants with metopic synostosis and additional anomalies, growth and developmental delay and recognition of the specific diagnosis can lead to important screening tests prior to planning surgery. For example, a patient presenting with metopic craniosynostosis and a concerning heart murmur could have a chromosome 11q deletion (Jacobsen syndrome). Infants with 11q deletion can have significant thrombocytopenia, congenital heart anomaly, abnormal brain structure, and developmental delays with medically actionable evaluations that would improve patient safety with surgery and overall care for the patient. Finally, ERF gene testing should be considered for individuals with non-syndromic multisuture craniosynostosis, especially if the lambdoid and sagittal sutures are involved and the synostosis has a progressive and postnatal onset, and in familial cases. Individuals with ERF variants are at risk of Chiari 1 malformation, increased intracranial pressure with risk of visual sequelae, low lying tonsils/hindbrain low-lying hernia, and global developmental delays.27,28,30,51–53
We propose that a flow chart (algorithm) based upon presenting clinical findings will help to identify of children who would benefit most from genetic consultation and testing and we have designed algorithms for three major groups of craniofacial conditions. These algorithms present an approach to genetic testing for the most common conditions managed by craniofacial teams: orofacial clefting, branchial arch conditions, and craniosynostosis. As outlined, timely diagnosis of genetic conditions is important, as it often leads to medical management recommendations that result in safer surgical interventions. Thus, these algorithms were designed to be applied at time of clinical presentation independent of age. Despite the high prevalence of craniofacial anomalies with a possible genetic etiology, these are the first comprehensive algorithms we are aware of for these conditions.
These algorithms were developed through a series of discussions with experts in the field from pediatrics, genetics, and genetic counseling, all of whom work with multidisciplinary craniofacial teams. We included several sites to decrease bias and broaden the perspective represented in this work. Furthermore, algorithms were presented in a 90-min workshop open to participants at the 2022 ACPA conference to improve consensus about the approaches to genetic testing. While reaching consensus among a broader group representing ACPA-accredited craniofacial teams or geneticists outside of ACPA was beyond the scope of this project and we recognize there may be some providers who choose a different approach to genetic testing, we believe these algorithms provide an approach that results in efficient and appropriate testing. Furthermore, the public sharing of these algorithms present an opportunity for a larger studies of their utility.
We recognize that the field of genetics is rapidly changing and these algorithms will need to be continuously updated. However, the approach presented here is intentionally general, suggesting when to recommend CMA or a panel (the latter of which could be modified as needed with updated identification of candidate genes). In addition to providing a framework for testing, we believe this approach will help any medical providers, whether in primary care settings or multidisciplinary craniofacial teams, determine who should be referred for a genetics evaluation. Adherence to this approach is likely to result in improved patient outcomes and better counseling for patients and families about recurrence risk and expected disease course. In the future, we would recommend prospective multicenter validation studies that include impact on patient management and outcome.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
IRB
This work was approved by the Institutional Review Board (IRB) at the University of California San Francisco. The IRB at University of Washington and the University of California Los Angeles determined this to be quality improvement work not requiring review. The IRB at the Children's Hospital of Los Angeles declined to review this retroactively.