
Abstract
Objective
To investigate the association between the sidedness of orofacial clefts and additional congenital malformations.
Design
Linkage of a national registry of cleft births to national administrative data of hospital admissions
Setting
National Health Service, England
Participants
2007 children born with cleft lip ± alveolus (CL ± A) and 2724 with cleft lip and palate (CLP) born between 2000 and 2012.
Main outcome measure
The proportion of children with ICD-10 codes for additional congenital malformations by the sidedness (left, right or bilateral) of orofacial clefts.
Results
For CL ± A phenotypes, there was no evidence for a difference in the prevalence of additional anomalies between left (22%, reference), right (22%, aOR 1.02, 95% CI 0.80 to 1.28; P = .90) and bilateral clefts (23%, aOR 1.09, 95% CI 0.75 to 1.57; P = .66). For CLP phenotypes, there was evidence of a lower prevalence of additional malformations in left (23%, reference) compared to right (32%, aOR 1.54, 95% CI 1.25 to 1.91; P < .001) and bilateral clefts (33%, aOR 1.64, 95% CI 1.35 to 1.99; P < .001).
Conclusions
The prevalence of additional congenital malformations was similar across sidedness subtypes with CL ± A phenotypes but was different for sidedness subtypes within CLP cases. These data support the hypothesis that CL ± A has a different underlying aetiology from CLP and that within the CLP phenotype, right sided CLP may lie closer in aetiology to bilateral CLP than it does to left sided CLP.
Background
There are consistent trends for sidedness in clefts of the lip with or without palate (CL ± P) that has been termed ‘directional asymmetry’.1,2 The prevalence of left unilateral CL ± P is twice that of right unilateral CL ± P.2,3 Bilateral CL ± P occurs less commonly than unilateral CL ± P, with a quoted prevalence ratio of 6:3:1 for left/right/bilateral sidedness, respectively. 4 Directional asymmetry is not a unique phenomenon to CL ± P, as left sided predominance is also observed in preauricular skin tags, congenital hip dysplasia and absent upper limb, whereas right sided predominance is seen in microtia and pre-auricular sinus. 5 Both genetic and environmental factors have been proposed in the aetiology of sidedness in CL ± P, yet mechanisms are currently poorly understood. 6
Orofacial clefts can occur in isolation or alongside additional congenital malformations (ACMs). Congenital malformations have been defined as defects involving a functional, structural, morphological or positional anomaly of a whole or part of a body organ system that tends to be macroscopic and occurs before birth. 7 ACMs can occur as part of a syndrome, which may arise due to a change within the genetic material (including single-gene disorders or larger chromosome anomalies), teratogenic exposures or disruption events during development. 8 However, the distinction between syndromic and non-syndromic clefts may not always be readily apparent at cleft diagnosis. Presently, consensus guidance is lacking both with regard to screening for additional malformations and genetic investigation for children born with orofacial clefts. The study and identification of co-occurring ACMs is therefore illuminating because it can give insights into the underlying aetiology of orofacial clefts, 9 and helps to inform the design of bespoke management pathways. 10
The United Kingdom (UK) is well positioned to investigate the association of CL ± P sidedness with ACMs due to the systematic collection of data on cleft at a nationwide level within a centralised healthcare system, which is free at the point of access. We have previously published the range and frequency of ACMs in 9403 children born with orofacial clefts and noted a difference in prevalence of ACMs between cleft lips ± alveolus (CL ± A) and cleft lip and palate (CLP). 11 This study aims to investigate whether there is any difference in the prevalence of ACMs by sidedness in both CL ± A and CLP using two national datasets in England, linked at the individual patient level.
Methods
Data Source
The study cohort was identified in the Cleft Registry and Audit NEtwork (CRANE) Database which is a national registry of all live-born children with an orofacial cleft (OFC) in the UK. Cases registered are most commonly identified antenatally or at birth (84%) and overall case ascertainment has recently been calculated as above 95%. 12 Children whose parents had given consent for their child's records to be linked to other datasets were eligible to be linked to the Hospital Episode Statistics (HES) database.
The HES database 13 contains records on all diagnoses and treatments made and given during admissions to National Health Service (NHS) hospitals in England. HES is derived from routine submission of data from each individual NHS hospital admission, primarily for the purposes of payment for and commissioning of healthcare in England. 14 Data is inputted locally by professional health coders before undergoing central data quality processing for validity and data cleaning checks prior to being published. The linked dataset contained records on births from January 1st 2000 up to December 31st 2012 and hospital admissions up to March 31st 2015, to allow sufficient time for the existence of ACMs to be identified and recorded.
Cleft Subtype Classification
Children born with either a unilateral or bilateral cleft lip with or without a cleft palate were included. In CRANE, cleft phenotypes and sidedness are categorised according to the LAHSAL classification (see supplementary Table 1). 15 In HES, cleft type was assigned according to the presence of selected procedure codes (Classification of Interventions and Procedures, OPCS 4.5) and/or diagnosis codes (tenth revision of the International Classification of Diseases, ICD-10) 16 in any of the available HES records. A stepwise, hierarchical approach was employed. First, the cleft reconstruction procedure codes (F03, F29, F32) were used to identify cleft type groups: CL ± A, CLP and cleft palate only. Second, the diagnosis code was used to distinguish between unilateral and bilateral cases. Children with cleft palate only and those whose cleft type was discordant between the two data sources were excluded from the study cohort.
Additional Congenital Malformations (ACMs)
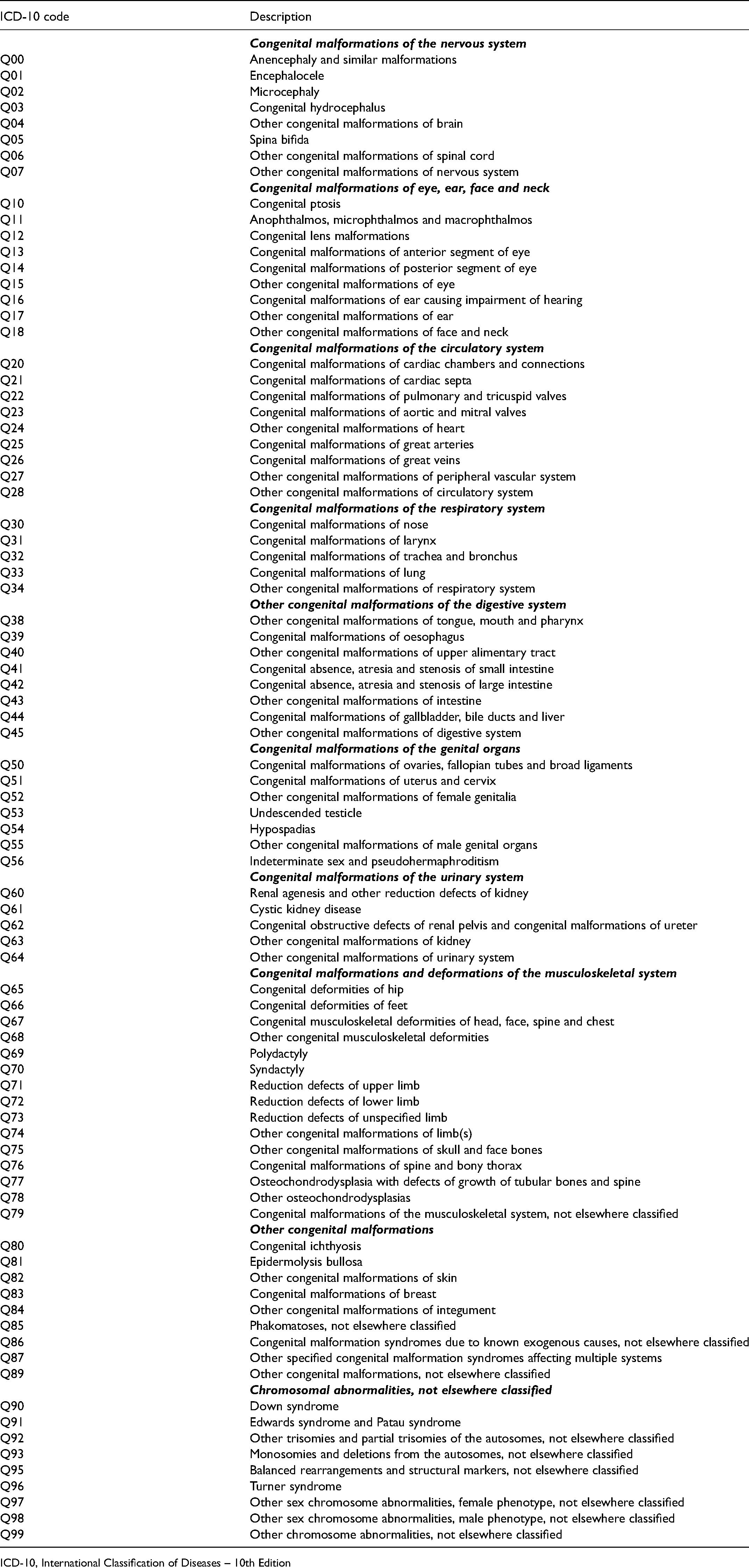
ACMs were classified based on the International Classification of Disease Version 10 (ICD-10) codes 16 which are reported in HES data. ICD-10 is considered the international standard diagnostic classification for disease and is developed and maintained by the World Health Organisation. Congenital malformations, deformations and chromosome abnormalities are coded Q00-Q99 and are categorised according to the body or organ system they affect (see Appendix 1). Codes for ACMs relating to cleft lip and palate (Q35-Q37) were excluded from the analysis. The code for nasal ACMs (Q30) was included in the analysis because despite the nose being positioned anatomically adjacent to the lip and palate, the specified nasal anomalies would not be expected to represent the underlying orofacial cleft (Q30.1 Agenesis and underdevelopment of nose; Q30.2 Fissured, notched and cleft nose; Q30.3 Congenital perforated nasal septum; Q30.8 Other congenital malformations of nose; Q30.9 Congenital malformation of nose, unspecified). Similarly, code Q38 relating to malformations of tongue, mouth and pharynx was included as it specified that it excluded cleft lip and/or palate.
We did not attempt to remove individuals with a specific genetic and/or syndromic diagnosis from the analysis cohort, since there was no means of reliably achieving this distinction using ICD-10 codes.
Data Analysis
The proportion of children with ICD-10 codes for congenital malformations (listed in Appendix 1) was examined. Differences in the percentage of children with ACMs between sidedness subgroups were initially explored using the Pearson chi-square test of independence. Children born with CL ± A were analysed separately from children born with CLP, as previous findings indicated different proportions of ACMs between the two phenotypes, 11 and in line with developing knowledge to suggest that these cleft subtypes should be considered as distinct aetiological entities.17–20
As an additional analysis, the number of different body or organ systems with malformations was summed for each child, and the percentage of children with ACMs affecting two or more body systems was reported by cleft subtype. ICD-10 codes Q90-Q99 (Chromosomal abnormalities not elsewhere specified) were not counted in this additional analysis as they do not relate to a particular body system.
The association between cleft sidedness (left, right or bilateral) and the presence of ACMs was analysed using logistic regression to estimate effect size, using left sided clefts as a reference group and adjusting for patient sex. Additional adjustment was made by adding ethnicity into the regression model, but due to incomplete ethnicity data reducing the sample size, these results are reported as supplementary data only. Odds ratios, confidence intervals and p values are reported and interpreted as continuous measures of the strength of evidence against the null hypothesis. 21 Statistical analysis was performed in Stata nV.17 (Statacorp).
Ethical Considerations
This study is exempt from NHS Research Authority ethics approval as it involves the analysis of an existing anonymised dataset that is collected for the purpose of service evaluation. 22
Results
Patient Characteristics
5940 CRANE-registered children were born alive in England with a CL ± P between 1st January 2000 and 31st December 2012. 5640 (95%) CRANE records were successfully linked to HES records but 1209 were excluded due to cleft phenotype discordance between the two data sources. The final cohort in this study included 4731 children born with CL ± P and of these, 42% had CL ± A and 58% had CLP (see Table 1). Among children with unilateral clefts, the ratio between left and right-sides was similar between those with CL ± A and those with CLP (65%:35% vs 63%:37% respectively, P = .279), However, the distribution of left, right and bilateral clefts differed between the CL ± A and CLP groups (59%, 32%, 9% vs. 43%, 25%, 32% respectively, P < .001).
Characteristics of the Children Included in the Analyses and the Number and Percentage of Those with ≥1 Additional Congenital Malformations (ACM).
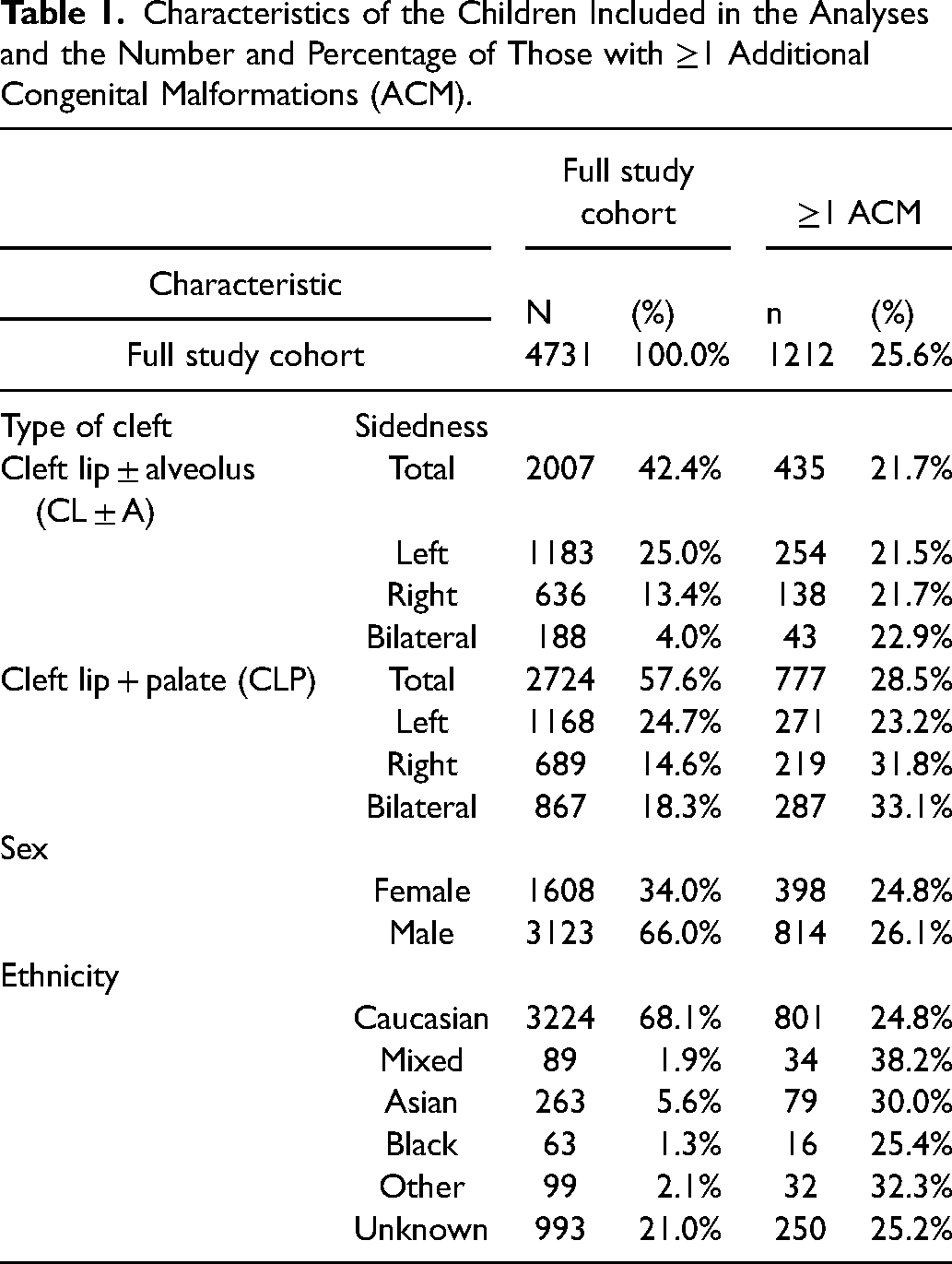
66% of the cohort was male. This proportion differed between CL ± A and CLP groups (63% vs 68%, P < .001) but there was no difference in the proportion of ACMs between males and females (26% vs 25% P = .33). Among those with reported ethnicity (79%), the proportion identified as Caucasian was similar between the CL ± A and CLP groups (87% vs 86%, P = .58). The overall proportion of ACMs was lowest in Caucasian and Black ethnicities and highest in Mixed, Asian and ‘Other’ ethnicity groups (see Table 1).
Additional Congenital Malformations (ACMS)
The prevalence of ACMs for the different cleft subtypes and sidedness are detailed in Tables 2 and 3 and Figures 1 and 2. When categorised by body systems, the two most prevalent ACMs for the CL ± A cohort were musculoskeletal (5%) and respiratory (5%), whereas for the CLP cohort they were circulatory (8%) and musculoskeletal (8%).
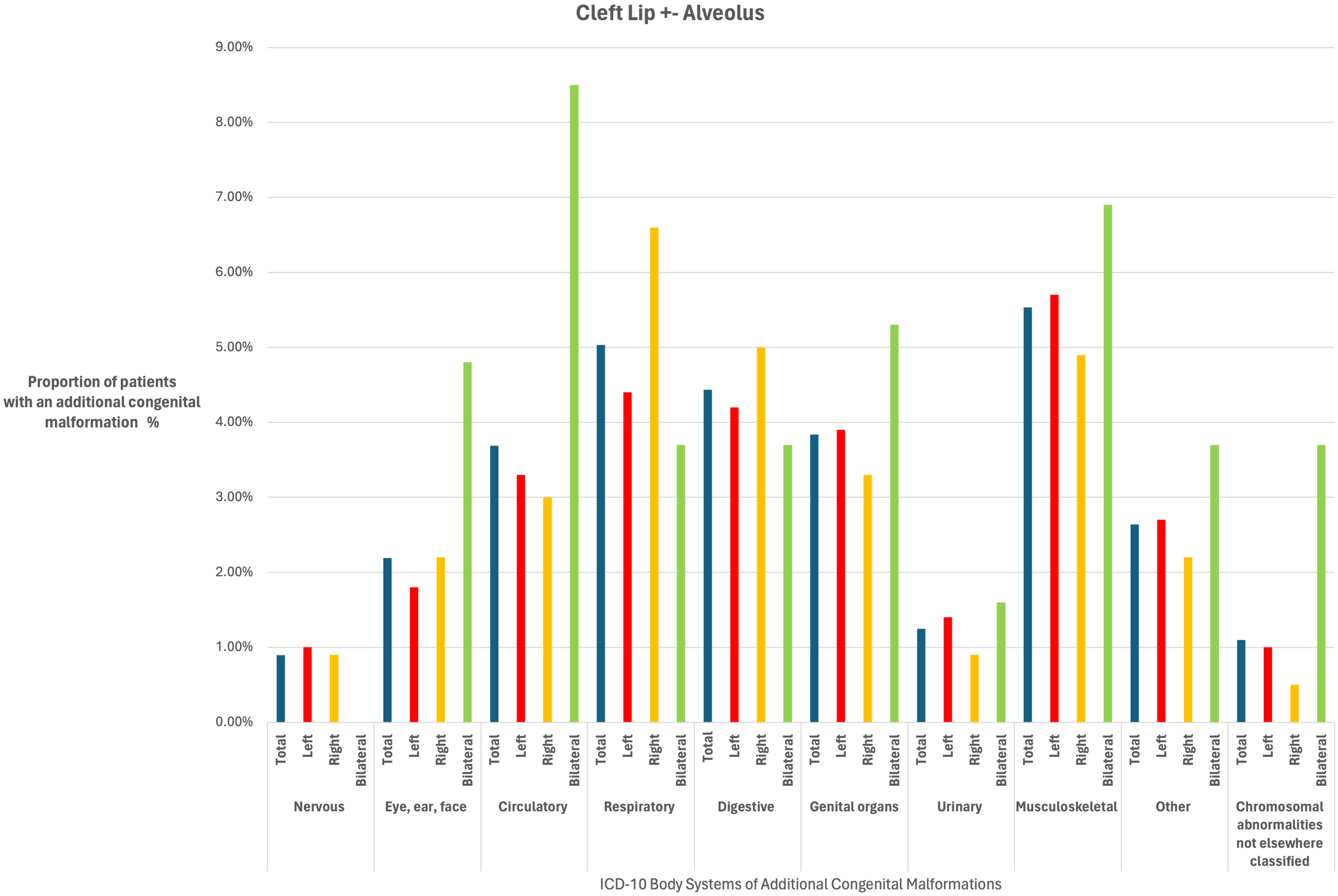
A bar chart to show the proportion (%) of patients born with a cleft lip ± alveolus with additional congenital malformations according to cleft sidedness (left, right and bilateral) for body systems in the International Classification of Disease Version 10 (ICD-10) system.
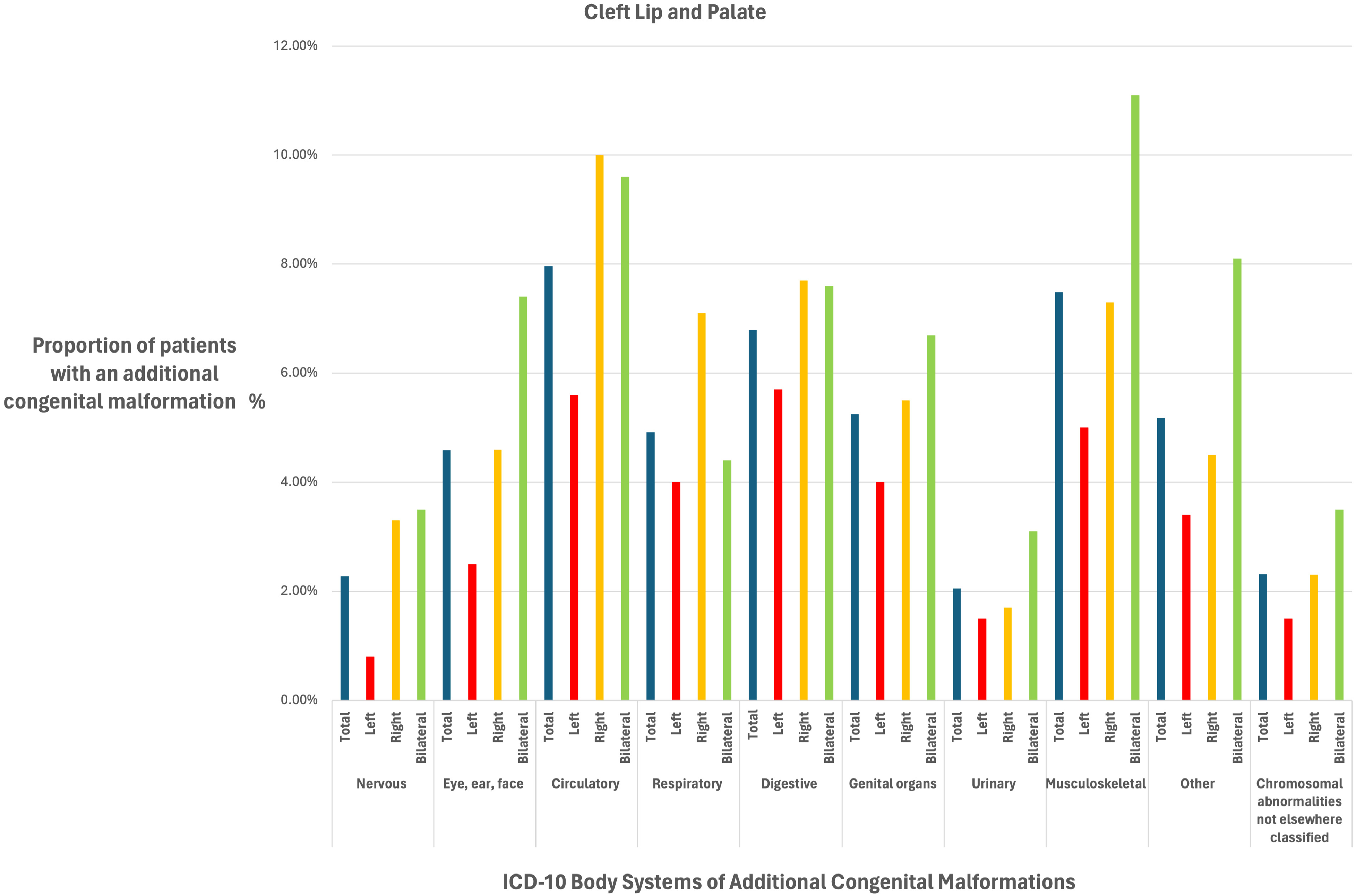
A bar chart to show the proportion (%) of patients born with a cleft lip + palate with additional congenital malformations according to cleft sidedness (left, right and bilateral) for body systems reported in the International Classification of Disease Version 10 (ICD-10) system.
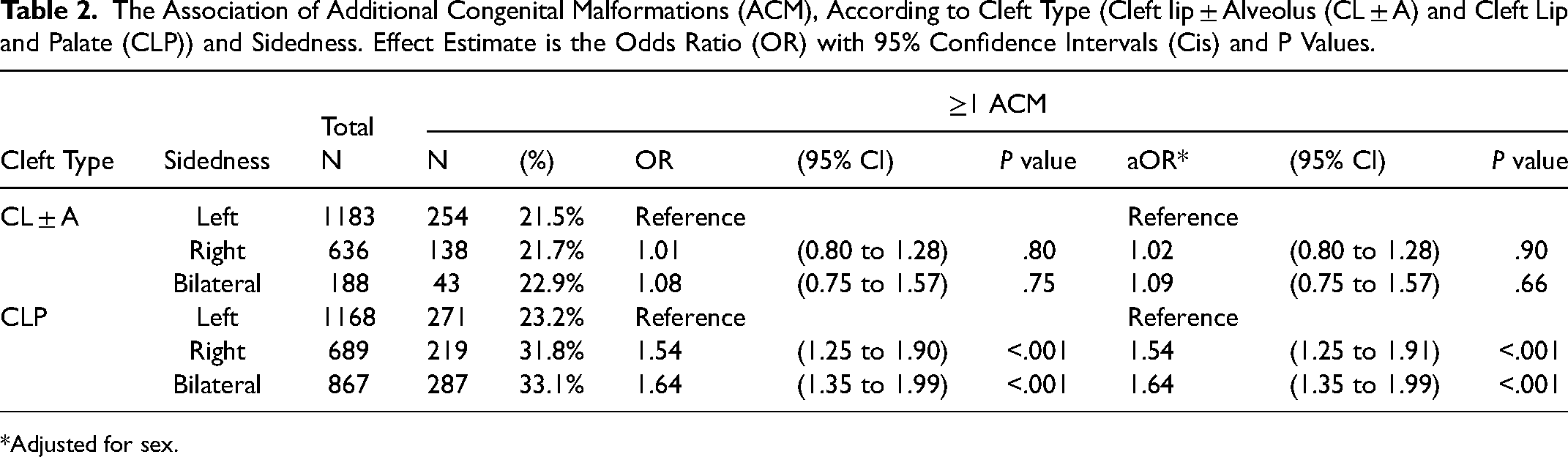
The Association of Additional Congenital Malformations (ACM), According to Cleft Type (Cleft lip ± Alveolus (CL ± A) and Cleft Lip and Palate (CLP)) and Sidedness. Effect Estimate is the Odds Ratio (OR) with 95% Confidence Intervals (Cis) and P Values.
Adjusted for sex.
The Association of Additional Congenital Malformations (ACM) According to ICD-10 Body System Classification, by Cleft Type (Cleft lip ± Alveolus (CL ± A) and Cleft Lip and Palate (CLP)) and Sidedness. Effect Estimate is the Odds Ratio (OR) with 95% Confidence Intervals (Cis) and P Values.
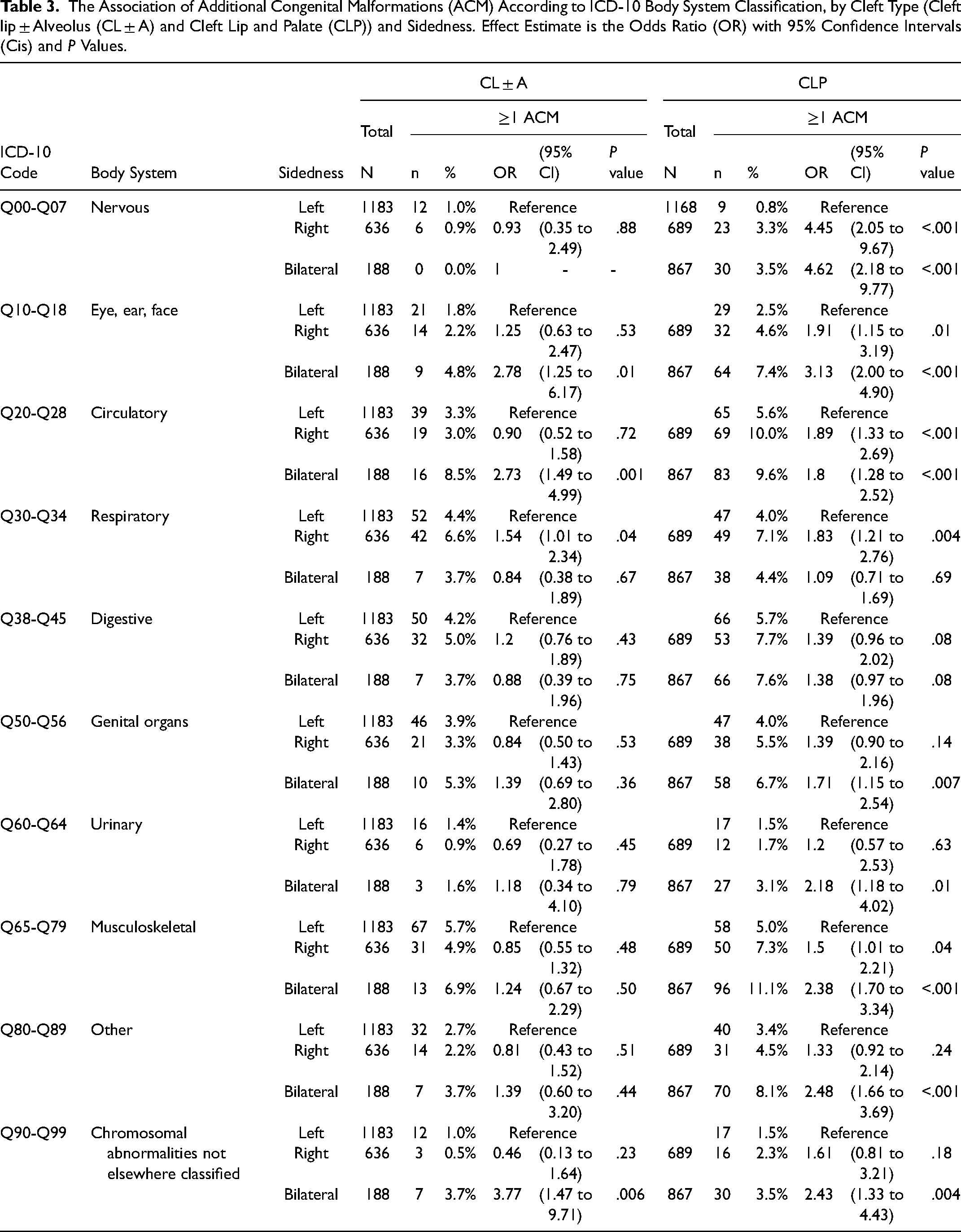
Cleft Lip ± Alveolus (CL ± A)
When compared to the proportion of ACMs in left sided CL ± A (22%), there was no difference in the proportion of ACMs in either right UCL ± A (22%, aOR 1.02, 95% CI 0.80 to 1.28; P = .90) or bilateral UCL ± A (23%, aOR 1.09, 95% CI 0.75 to 1.57; P = .66) as seen in Table 2. Additional adjustment for ethnicity did not alter this finding (see Supplementary Table 2).
When ACMs were stratified by the ICD-10 body system subgroups, the ten subcategories were present in similar proportions among children with a left, right or bilateral CL ± A in many of the body systems (See Table 3). When compared to the left CL ± A reference group, respiratory anomalies were more common in children with a right CL ± A (OR 1.54, 95% CI 1.01 to 2.34; P = .04), whereas the bilateral CL ± A phenotype had a higher proportion of eye, ear, face anomalies (OR 2.78, 95% CI 1.25 to 6.17; P = .01) and circulatory anomalies (OR 2.73, 95% CI 1.49 to 4.99; P = .001), although the evidence for these associations was weak given that we performed multiple tests.
Cleft Lip and Palate (CLP)
When compared to the proportion of ACMs in left sided CLP (23%) there was a higher proportion of ACMs amongst both right CLP (32%, aOR 1.54, 95% CI 1.25 to 1.91; P < .001) and bilateral CLP (33%, aOR 1.64, 95% CI 1.35 to 1.99; P < .001) as seen in Table 2. Additional adjustment for ethnicity did not alter this trend (Supplementary Table 2).
When ACMs were stratified by the ICD-10 body system subgroups, each of the ten subcategories of ACMs were more prevalent in both right and bilateral CLP when compared to the left CLP reference (see Table 3) The direction of the effect estimate for right and bilateral clefts was consistent for each body system, but the strength of association for ACMs compared to left CLP varied depending on the body system.
Number of Systems Affected by Additional Congenital Malformations
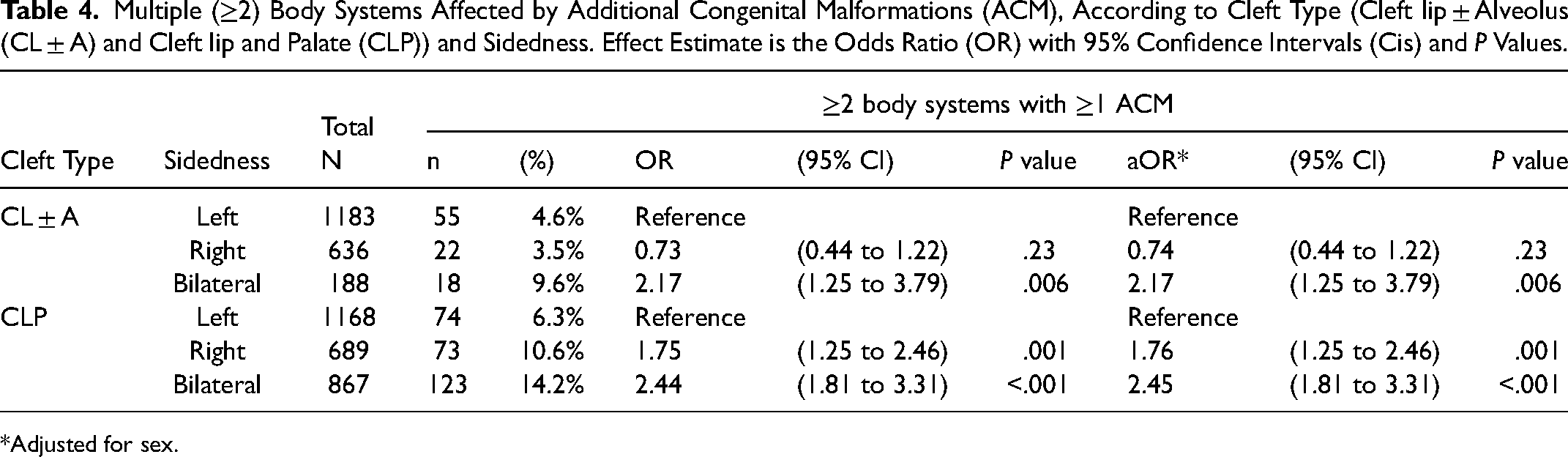
Table 4 shows the number and percentage of children who had multiple (≥2) body systems (eg, nervous, eye/ear/face, circulatory, respiratory, digestive, genital, urinary and musculoskeletal systems) affected by ACMs. Among those with CL ± A, the proportion of children who had malformations across multiple body systems when compared to left CL ± A (5%) was similar for right CL ± A (4%, aOR 0.74, 95% CI 0.44 to 1.22; P = .23) but was greater for bilateral CL ± A (10%, aOR 2.17, 95% CI 1.25 to 3.79; P < .001). Among those with CLP, the proportion of children who had ACMs across multiple body systems when compared to left CLP (6%), was greater for both right CLP (11%, aOR 1.76, 95% CI 1.25 to 2.46; P < .001) and bilateral CLP (14%, aOR 2.45, 95% CI 1.81 to 3.31; P < .01).
Multiple (≥2) Body Systems Affected by Additional Congenital Malformations (ACM), According to Cleft Type (Cleft lip ± Alveolus (CL ± A) and Cleft lip and Palate (CLP)) and Sidedness. Effect Estimate is the Odds Ratio (OR) with 95% Confidence Intervals (Cis) and P Values.
Adjusted for sex.
Discussion
Summary of Findings
In this national sample of 4731 children born with CL ± P, we were able to examine the association of cleft sidedness with the prevalence of ACM as coded by the ICD-10 classification. The prevalence of ACMs for children born with CL ± A ranged between 22% to 23% and whilst there was no evidence to suggest differences between left, right or bilateral sidedness, there was evidence to suggest a greater proportion of ACMs in multiple body systems in bilateral CL ± A (10%) compared to left (5%) or right (4%) CL ± A.
The prevalence of ACMs for children born with CLP was different according to sidedness, with 23% of left sided CLP having at least one additional malformation compared to 32% in right CLP and 33% in bilateral CLP. The subcategorization of ICD-10 malformation codes into body systems within the CLP subtype, consistently showed a higher prevalence of malformations for right and bilateral CLP compared to left CLP in each body system, reducing the likelihood that this trend is a chance finding. In addition, the proportion of ACMs in multiple body systems was greater for both right (11%) and bilateral (14%) CLP compared to left CLP (6%).
Comparison to Previous Studies
Pereira et al., 10 analysed the presence of ACMs in 701 children born with orofacial clefts from a Portuguese database, 343 of whom had unilateral CL ± P. Among children with ACMs, there was a higher prevalence of right CL ± P compared to the group without ACMs (13% vs 11%; P < .001). The authors did not stratify individual ACMs by sidedness but, for the entire cohort, head and neck anomalies were found to be the most common, followed by cardiovascular anomalies.
Gallagher et al., 23 reported a greater prevalence of syndromes amongst 155 children born with a right sided CL ± P compared to 276 born with a left sided CL ± P (OR 1.9, 95% CI 0.7 to 4.7). The imprecise estimate may have been linked to the small sample size and subjective method of identifying syndromes. The authors concluded that children born with right sided CL ± P were more likely to be born with ACMs and whilst this is consistent with our data, they did not subcategorise the clefts into CL ± A and CLP subtypes.
Interpretation of the Data
Our data can be used to consider the current theories that have been suggested for the aetiology of sidedness in CL ± P. Various case control and genome wide association studies (GWAS) have highlighted distinct genetic variations associated with left versus right sided CL ± P over the past two decades.20,24–28 For example, using a GWAS approach, Curtis et al., 20 found evidence that a polymorphism at locus 4q28, close to the FAT4 gene, influenced sidedness in unilateral CL ± A, whilst no significant effect was detected for this locus in unilateral CLP. For decades, since pioneering work by Fogh-Anderson, 29 it has been recognised that the aetiology of cleft palate only is likely different to that of CL ± P. Further, subsequent evidence has emerged to suggest distinct aetiologies between CL ± A and CLP. 17 Our data contributes further evidence that sidedness may also be distinct between CL ± A and CLP subtypes.
The bilateral CL ± P cohort in this study provides a useful comparative group for the unilateral CL ± P cohort. First, cleft lip sidedness ratios are often quoted in the literature as 6:3:1 for left:right:bilateral 4 and whilst this was true in our CL ± A cohort, the proportion of bilateral CLP was similar to right CLP, as has been previously reported in other orofacial cleft epidemiology studies.2,30 Second, for the CL ± A subtype, the prevalence of ACMs is similar irrespective of left, right or bilateral sidedness, whereas for the CLP subtype the prevalence of ACMS is higher for right and bilateral CLP compared to left CLP.
The amalgamation of all cleft subtypes included in our main and supplementary analysis can be presented as a spectrum using left CL ± A as a reference (see Figure 3 and supplementary Table 3) and demonstrates right and bilateral CLP standing apart from the other cleft lip subtypes, according to the proportion of associated ACMs. In two studies investigating the co-occurrence of congenital dental anomalies with cleft sidedness, there was a reported higher prevalence of left maxillary lateral incisor agenesis in right CL/P compared with the reverse scenario.31,32 Both authors suggested the occurrence of a missing lateral incisor on the non-cleft side may represent a bilateral genotype that did not fully manifest and that right sided CL ± P may be more likely to correspond to a milder phenotype of bilateral clefts.33,34
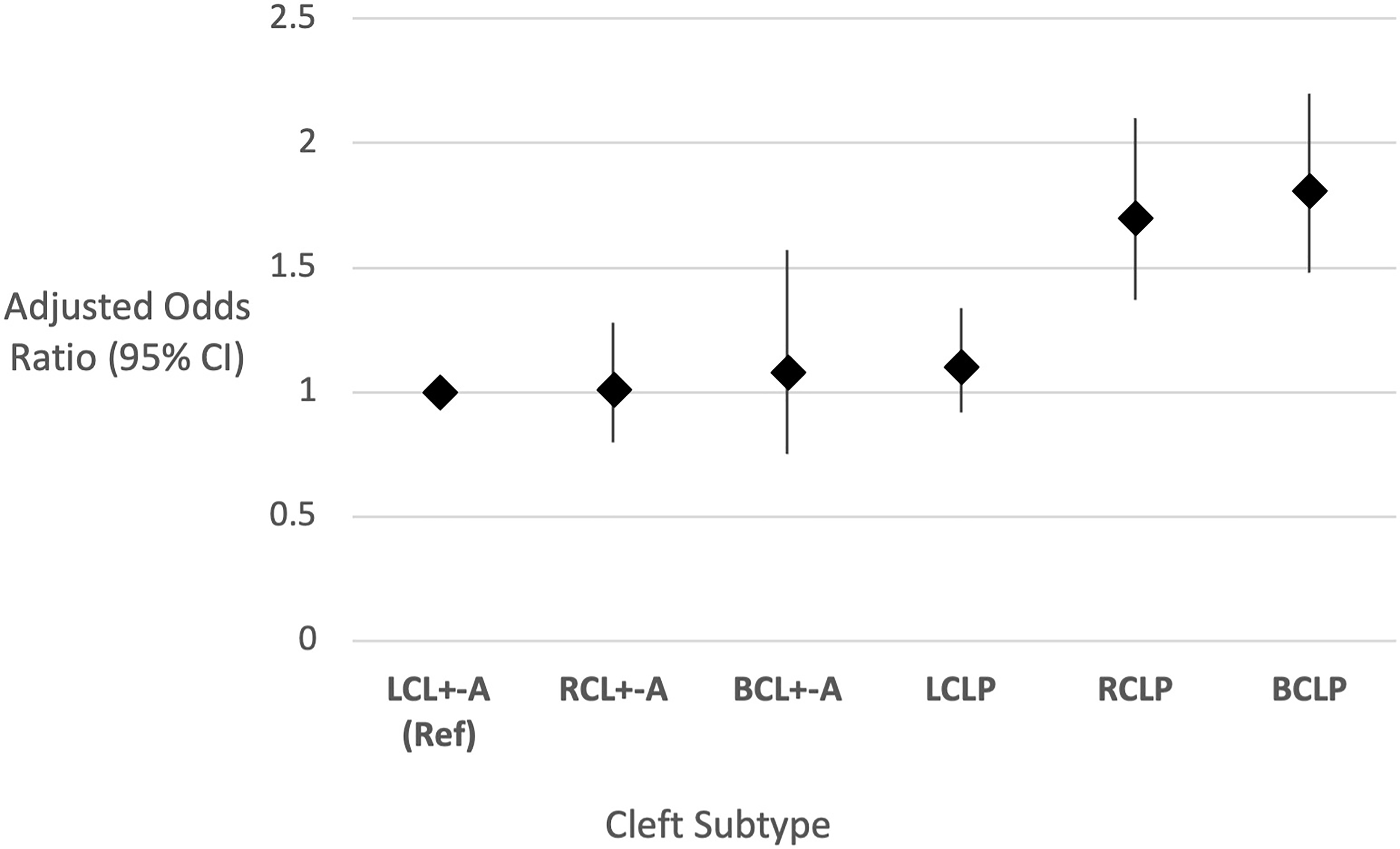
The association of additional congenital malformations across the spectrum of unilateral and bilateral cleft lip with or without cleft palate. Groups are categorised according to sidedness left (L), right (R) and bilateral (B) and the cleft subtypes cleft lip ± alveolus (CL ± A) and cleft lip and palate (CLP). Left unilateral cleft lip ± A (LUCL ± A) is used as the reference group. Effect estimates displayed as Odds Ratio (OR) with 95% Confidence Intervals (CI), adjusted for sex.
Investigations into the aetiology of orofacial cleft have reached a broad consensus that non-syndromic clefts arise within a multifactorial background.33,34 According to the Multifactorial threshold Model of Inheritance, right sided CLP and bilateral CLP occur less commonly than left sided CLP, requiring a higher threshold of genetic and environmental influences for them to occur and may represent more severe phenotypes.
Strengths and Limitations
This is a large cohort of standardised national data within a centralised and nationalised health care system, and therefore represents a unique resource. The CRANE registry has high case ascertainment, 12 and given its national coverage is likely to be representative of children born with an orofacial in England during the study period. A minimum follow-up of 2.25 years in HES data allowed sufficient time for congenital anomalies to be identified after birth. Although the cohort was born between 2000 and 2012, we believe that the knowledge gained from this study is still relevant to people born with an orofacial cleft in the present day. Discordance in cleft subtypes in one fifth of the patient records between the datasets was a weakness as it resulted in the exclusion of otherwise successfully linked data.
The ICD-10 categorisation system of malformations is advantageous because it is an international system that is widely accessible and facilitates thorough documentation of anatomical detail. However, despite the ICD-10 being regarded as the international standard, a variety of different classifications have been used in the literature, which makes comparison difficult.10,35 The ICD-10 anatomical subcategorization is not necessarily clinically intuitive and cannot differentiate between minor and major ACMs. Whilst the use of ICD-10 codes allowed us to report many congenital malformations, HES restricts the entry of these codes to 4 characters (eg, Q87.0), which means that some codes were not sensitive enough to distinguish between certain diagnoses (for example, Pierre Robin Sequence and Goldenhar syndrome share the same 4-character ICD-10 code). Furthermore, ICD-10 codes utilised in HES tend to focus on a physical diagnosis or phenotype, rather than the underlying genetic cause. This means the prevalence of specific genetic and/or syndromic diagnoses associated with orofacial clefts and other congenital malformations could not be reported. With the now widespread application of genomic testing, this limitation highlights a need for cleft research cohorts to accurately capture data relating to genetic and syndromic diagnoses.
This study was restricted to children born alive. It was not possible to include spontaneous abortions, elective terminations and stillborn foetuses. Furthermore, as there is no standard protocol for evaluating other body systems for anomalies in children presenting with a cleft in England, there may well be subclinical and untreated anomalies that have been missed in the study population. Whilst there is no reason to suggest that the prevalence of missed ACMs would differ according to cleft sidedness, the true prevalence of ACMs is likely to be underrepresented.
Finally, we adjusted for the potential confounders of sex and ethnicity as these variables were readily available in the data set. Adjustment for these confounders did not alter the trends observed indicating that the reported findings were not explained by these characteristics. The proportion of children with additional malformations according to ethnic background was limited by missing data and slightly lower representation of minority ethnic groups in this cohort as compared to last UK census. 36 Furthermore, we were not able to add additional confounding factors to our model, which could be important in cleft aetiology, such as maternal age, Body Mass Index, and folic acid consumption.
Further Work
Our study demonstrates the importance of any study on cleft sidedness being explicit about the inclusion or exclusion of children born with ACMs, as the proportion of ACMs will likely be different for children born with left, right and bilateral CLP. Further work to understand the aetiology of sidedness in CL ± P should stratify participants by CL ± A and CLP subtypes. Furthermore, future studies should make efforts to distinguish individuals within cleft cohorts who have a known monogenic or chromosomal diagnosis, from those whose cleft is assumed to have a complex or multifactorial cause, since genetic influences may be overlapping or distinct between these groups.
Our results suggest a research focus on left CLP versus right and bilateral CLP is warranted. Work to investigate genetic factors should be expanded to include large scale GWAS studies. DNA methylation has been shown to play a critical role in establishing laterality of the body plan during early embryogenesis in zebrafish models, 37 and methylation patterns have been shown to differ by CL ± A, CLP and CP subtype in humans, 17 yet we are not aware of its use in orofacial cleft sidedness research and studying methylation patterns could offer important insights in this area. This may help to enhance our understanding of how cleft sub-phenotypes are classified and provide more information to support or refute the suggestion of right sided CLP and bilateral CLP being different phenotypic expressions of the same condition.
Conclusion
This large population-based study provides evidence for an increased risk of ACMs among right and bilateral CLP compared to left CLP. This was not the case for CL ± A, where ACMs were equivalent according to cleft sidedness. This supports a growing evidence base to suggest CLP has a different aetiology when compared with CL ± A and that these subtypes of cleft should not be considered as a single entity. Furthermore, this data suggests that right CLP may lie closer in aetiology to bilateral CLP than it does to left sided CLP. Our findings may provide further insights into why right sided orofacial clefts are less prevalent than left sided orofacial clefts, although further work is required to explain, define and understand this.
Supplemental Material
sj-docx-1-cpc-10.1177_10556656241261918 - Supplemental material for Cleft lip Sidedness and the Association with Additional Congenital Malformations
Supplemental material, sj-docx-1-cpc-10.1177_10556656241261918 for Cleft lip Sidedness and the Association with Additional Congenital Malformations by Matthew Fell, Kate J. Fitzsimons, Mark J. Hamilton, Jibby Medina, Sophie Butterworth, Min Hae Park, Jan Van der Meulen, Sarah Lewis, David Chong and Craig JH Russell in The Cleft Palate Craniofacial Journal
Supplemental Material
sj-docx-2-cpc-10.1177_10556656241261918 - Supplemental material for Cleft lip Sidedness and the Association with Additional Congenital Malformations
Supplemental material, sj-docx-2-cpc-10.1177_10556656241261918 for Cleft lip Sidedness and the Association with Additional Congenital Malformations by Matthew Fell, Kate J. Fitzsimons, Mark J. Hamilton, Jibby Medina, Sophie Butterworth, Min Hae Park, Jan Van der Meulen, Sarah Lewis, David Chong and Craig JH Russell in The Cleft Palate Craniofacial Journal
Supplemental Material
sj-docx-3-cpc-10.1177_10556656241261918 - Supplemental material for Cleft lip Sidedness and the Association with Additional Congenital Malformations
Supplemental material, sj-docx-3-cpc-10.1177_10556656241261918 for Cleft lip Sidedness and the Association with Additional Congenital Malformations by Matthew Fell, Kate J. Fitzsimons, Mark J. Hamilton, Jibby Medina, Sophie Butterworth, Min Hae Park, Jan Van der Meulen, Sarah Lewis, David Chong and Craig JH Russell in The Cleft Palate Craniofacial Journal
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Sarah Lewis is supported by the Medical Research Council, (grant number MR/T002093/1).
Supplemental Material
Supplemental material for this article is available online.
Appendix 1: International Classification of Disease Version 10 (ICD-10) diagnostic codes used to identify congenital malformations in Hospital Episode Statistics.
| ICD-10 code | Description |
|---|---|
|
|
|
| Q00 | Anencephaly and similar malformations |
| Q01 | Encephalocele |
| Q02 | Microcephaly |
| Q03 | Congenital hydrocephalus |
| Q04 | Other congenital malformations of brain |
| Q05 | Spina bifida |
| Q06 | Other congenital malformations of spinal cord |
| Q07 | Other congenital malformations of nervous system |
|
|
|
| Q10 | Congenital ptosis |
| Q11 | Anophthalmos, microphthalmos and macrophthalmos |
| Q12 | Congenital lens malformations |
| Q13 | Congenital malformations of anterior segment of eye |
| Q14 | Congenital malformations of posterior segment of eye |
| Q15 | Other congenital malformations of eye |
| Q16 | Congenital malformations of ear causing impairment of hearing |
| Q17 | Other congenital malformations of ear |
| Q18 | Other congenital malformations of face and neck |
|
|
|
| Q20 | Congenital malformations of cardiac chambers and connections |
| Q21 | Congenital malformations of cardiac septa |
| Q22 | Congenital malformations of pulmonary and tricuspid valves |
| Q23 | Congenital malformations of aortic and mitral valves |
| Q24 | Other congenital malformations of heart |
| Q25 | Congenital malformations of great arteries |
| Q26 | Congenital malformations of great veins |
| Q27 | Other congenital malformations of peripheral vascular system |
| Q28 | Other congenital malformations of circulatory system |
|
|
|
| Q30 | Congenital malformations of nose |
| Q31 | Congenital malformations of larynx |
| Q32 | Congenital malformations of trachea and bronchus |
| Q33 | Congenital malformations of lung |
| Q34 | Other congenital malformations of respiratory system |
|
|
|
| Q38 | Other congenital malformations of tongue, mouth and pharynx |
| Q39 | Congenital malformations of oesophagus |
| Q40 | Other congenital malformations of upper alimentary tract |
| Q41 | Congenital absence, atresia and stenosis of small intestine |
| Q42 | Congenital absence, atresia and stenosis of large intestine |
| Q43 | Other congenital malformations of intestine |
| Q44 | Congenital malformations of gallbladder, bile ducts and liver |
| Q45 | Other congenital malformations of digestive system |
|
|
|
| Q50 | Congenital malformations of ovaries, fallopian tubes and broad ligaments |
| Q51 | Congenital malformations of uterus and cervix |
| Q52 | Other congenital malformations of female genitalia |
| Q53 | Undescended testicle |
| Q54 | Hypospadias |
| Q55 | Other congenital malformations of male genital organs |
| Q56 | Indeterminate sex and pseudohermaphroditism |
|
|
|
| Q60 | Renal agenesis and other reduction defects of kidney |
| Q61 | Cystic kidney disease |
| Q62 | Congenital obstructive defects of renal pelvis and congenital malformations of ureter |
| Q63 | Other congenital malformations of kidney |
| Q64 | Other congenital malformations of urinary system |
|
|
|
| Q65 | Congenital deformities of hip |
| Q66 | Congenital deformities of feet |
| Q67 | Congenital musculoskeletal deformities of head, face, spine and chest |
| Q68 | Other congenital musculoskeletal deformities |
| Q69 | Polydactyly |
| Q70 | Syndactyly |
| Q71 | Reduction defects of upper limb |
| Q72 | Reduction defects of lower limb |
| Q73 | Reduction defects of unspecified limb |
| Q74 | Other congenital malformations of limb(s) |
| Q75 | Other congenital malformations of skull and face bones |
| Q76 | Congenital malformations of spine and bony thorax |
| Q77 | Osteochondrodysplasia with defects of growth of tubular bones and spine |
| Q78 | Other osteochondrodysplasias |
| Q79 | Congenital malformations of the musculoskeletal system, not elsewhere classified |
|
|
|
| Q80 | Congenital ichthyosis |
| Q81 | Epidermolysis bullosa |
| Q82 | Other congenital malformations of skin |
| Q83 | Congenital malformations of breast |
| Q84 | Other congenital malformations of integument |
| Q85 | Phakomatoses, not elsewhere classified |
| Q86 | Congenital malformation syndromes due to known exogenous causes, not elsewhere classified |
| Q87 | Other specified congenital malformation syndromes affecting multiple systems |
| Q89 | Other congenital malformations, not elsewhere classified |
|
|
|
| Q90 | Down syndrome |
| Q91 | Edwards syndrome and Patau syndrome |
| Q92 | Other trisomies and partial trisomies of the autosomes, not elsewhere classified |
| Q93 | Monosomies and deletions from the autosomes, not elsewhere classified |
| Q95 | Balanced rearrangements and structural markers, not elsewhere classified |
| Q96 | Turner syndrome |
| Q97 | Other sex chromosome abnormalities, female phenotype, not elsewhere classified |
| Q98 | Other sex chromosome abnormalities, male phenotype, not elsewhere classified |
| Q99 | Other chromosome abnormalities, not elsewhere classified |
ICD-10, International Classification of Diseases – 10th Edition
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.