
Abstract
Cranial sutures are complex structures integrating mechanical forces with osteogenesis which are often affected in craniofacial syndromes. While premature fusion is frequently described, rare pathological widening of cranial sutures is a comparatively understudied phenomenon. This narrative review aims to bring to light the biologically variable underlying causes of widened sutures and persistent fontanelles leading to a common outcome. The authors herein present four syndromes, selected from a literature review, and their identified biological mechanisms in the context of altered suture physiology, exploring the roles of progenitor cell differentiation, extracellular matrix production, mineralization, and bone resorption. This article illustrates the gaps in understanding of complex craniofacial disorders, and the potential for further unification of genetics, cellular biology, and clinical pillars of health science research to improve treatment outcomes for patients.
Introduction
Cranial sutures, the fibrous joints connecting skull bones, have been the object of craniofacial study for millennia, with the earliest categorization of their number and arrangement dating back to the Hippocratic era. 1 Since these early morphological descriptions, the function of cranial sutures as secondary growth zones of calvaria has been uncovered. 2 The cranial vault is composed of paired frontal, parietal, and temporal bones, and a single posterior occipital bone. These membranous bones, called calvaria, are separated by cranial sutures, which intersect each other at fontanelles.
Critical for expansion of the skull throughout development, sutures represent a complex system in which mechanical forces interface with osteogenesis. Suture patency is mainly governed by stem and progenitor cells residing within the suture, which differentiate along the intramembranous ossification pathway toward bone-forming osteoblasts that line the bone (Figure 1). 3 Other factors influence suture development and function, such as signals and cells from the dura mater and periosteum. 4 Bone and stem cell biology thus weave into the cranial suture system, and many suture malformations can be traced to abnormalities in key gene regulatory networks and processes.
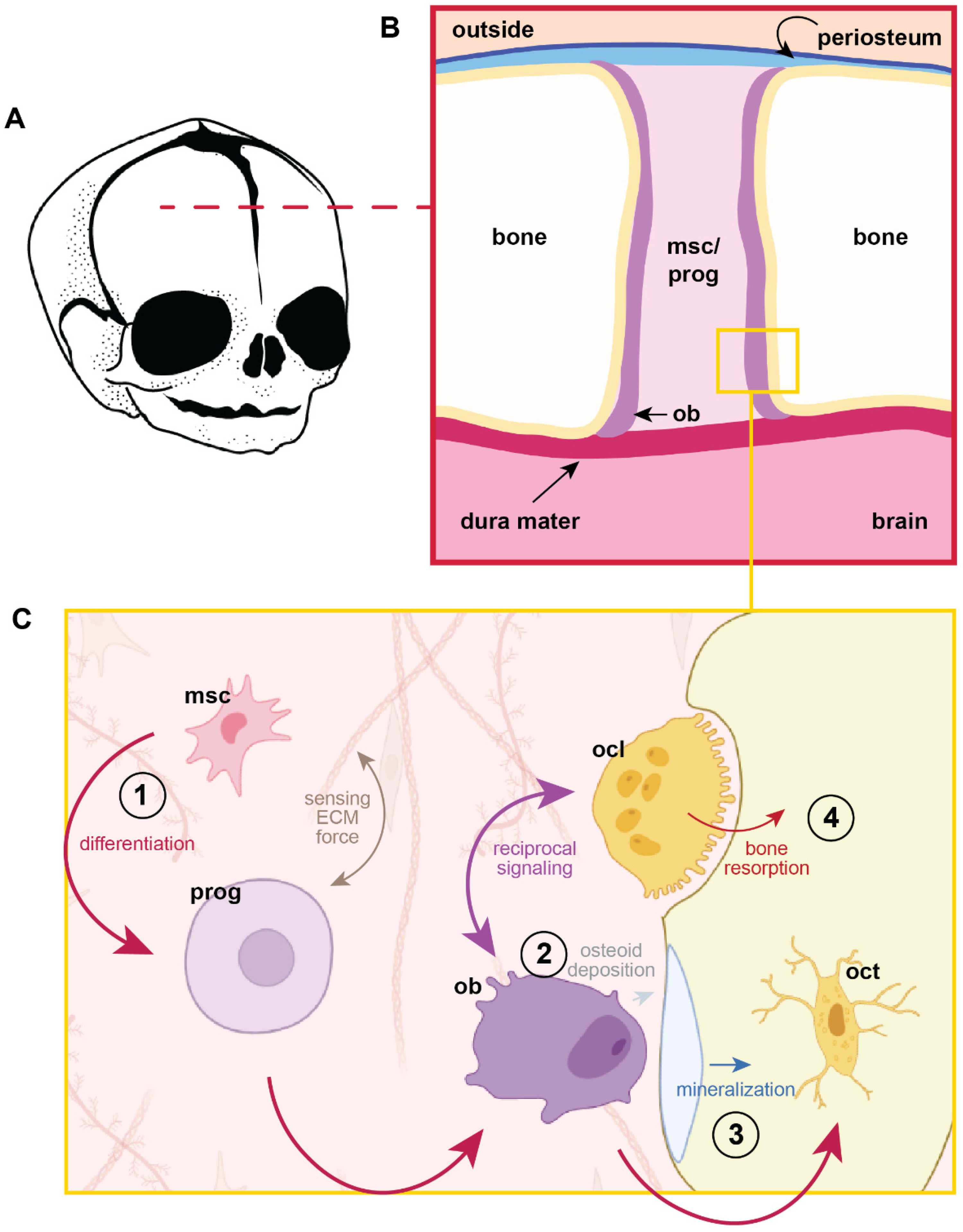
Cranial suture milieu.
The clinical significance of cranial sutures is mainly attributed to abnormalities in their intrinsic growth potential. Normal sutures and fontanelles fuse according to a predictable timeline throughout development; the anterior fontanelle ossifies in children around 10 months of age.5,6 Suture-related disorders involve either reduced or overactive bone formation within this niche, with major overall physiological implications. Craniosynostosis, for example, is a result of premature obliteration of the cranial sutures by bone, preventing appropriate expansion of the skull to accommodate mounting intracranial pressures with brain development. Common outcomes of craniosynostosis are facial and skull deformities and a host of neurological symptoms, such as raised intracranial pressure, seizures, developmental delay, and behavioral effects often accompanied by failure to thrive, though cause and consequence can be difficult to separate in syndromic cases. 7
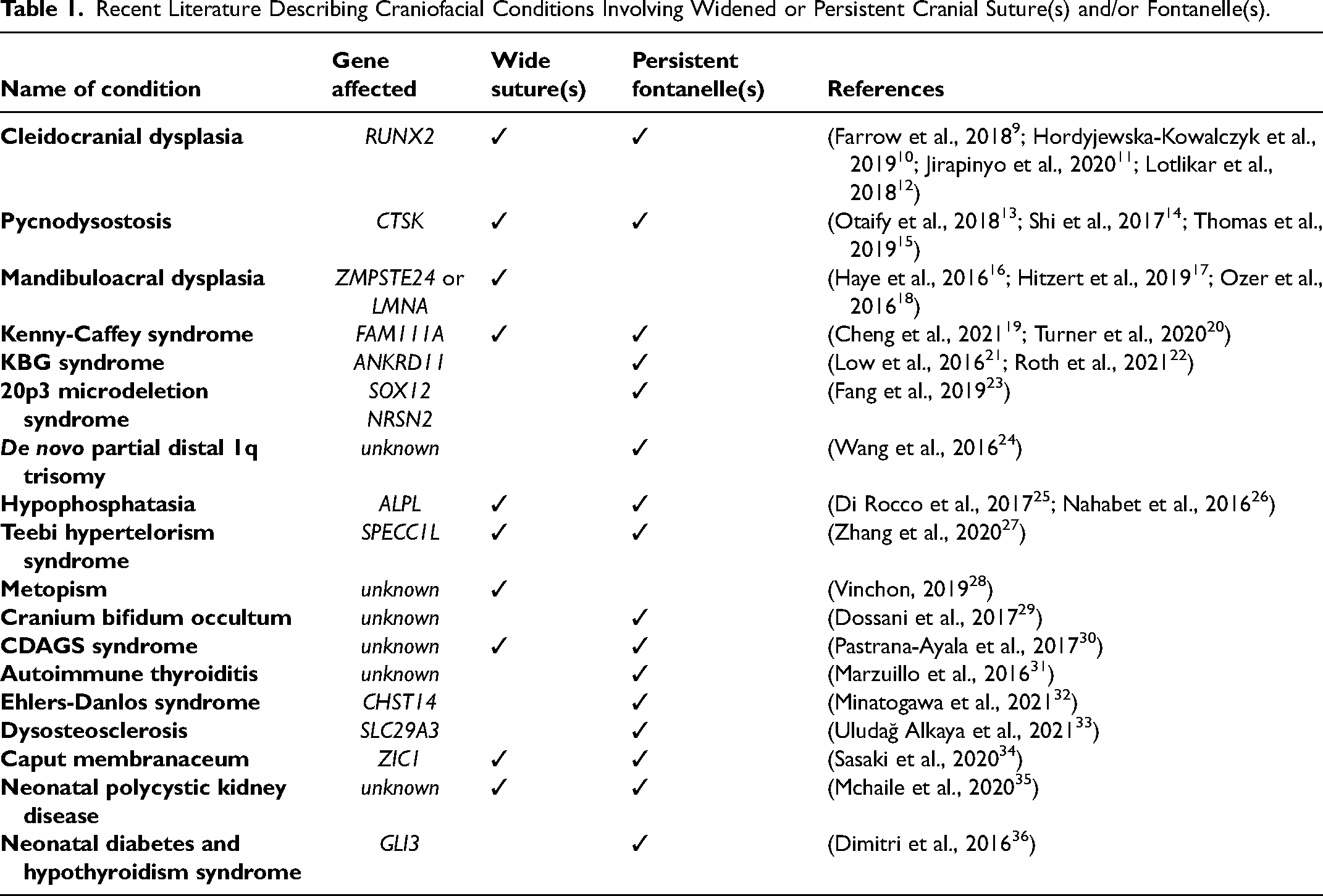
The craniosynostotic skull and its bony sutures have attracted significant study of cranial suture malformation and treatment modalities to return or enhance obliterated suture function. 8 A comparatively understudied phenomenon is the widening and persistence of the cranial suture space, seen clinically in several rare bone disorders (Table 1). This narrative review aims to present exemplary cases of different causes for similarly widened cranial sutures due to defects in progenitor cell differentiation (cleidocranial dysplasia), extracellular matrix production (Yunis-Varón syndrome), mineralization (hypophosphatasia), and bone resorption (pycnodysostosis) selected from recent reports in the literature. These examples highlight the importance of early, precise diagnosis to determine the correct treatment modality, bearing in mind potentially overlapping clinical signs. While sutures and fontanelles are not necessarily the same structure, for simplicity we consider expanded or abnormally persistent fontanelles to be an extension of, or variation on, widened cranial sutures.
Recent Literature Describing Craniofacial Conditions Involving Widened or Persistent Cranial Suture(s) and/or Fontanelle(s).
Methods
The disorders discussed in this narrative review were identified through a targeted literature search in PubMed using the following search terms: (((“delayed closure” OR wide* OR dysplasia OR persistent) AND (“cranial suture*” OR “fontanelle”)) AND (patent)) AND ((“2016/01/01"[Date—Publication]: “2022/01/01"[Date—Publication])). 90 results were obtained over the 7-year period. Of those, 28 articles were considered relevant to the topic of this article based on abstract review. These studies were reviews or case reports describing 18 conditions that manifest with widened sutures and/or persistent fontanelles, summarized in Table 1. Subsequent un-restricted literature search conducted in PubMed, Web of Science, and Ovid databases targeted pertinent cranial suture descriptions, etiology, and findings from the clinic and experimental systems based on the disorders identified in the preliminary search. The four clinical examples investigated in this review were chosen to illustrate independent biological principles of suture malformation.
Biologically Contextualized Clinical Examples
Cranial suture development and function are governed by distinct biological processes, including progenitor cell differentiation, extracellular matrix production and maturation, mineralization, and bone remodeling and resorption. The following clinical examples of suture widening are organized by these key processes in suture biology, illustrating that distinct underlying mechanisms can result in convergence of clinical phenotypic presentation.
Progenitor Cell Differentiation–Cleidocranial Dysplasia
Cleidocranial dysplasia (CCD) is a skeletal dysplasia with a frequency of one in one million individuals worldwide. 37 Clinically, individuals with CCD present with a triad of delayed closure of the cranial sutures and the anterior fontanelle, hypoplastic or aplastic clavicles, and dental abnormalities. Other features include delayed ossification of the skull, maxillary hypoplasia, moderate short stature, and other skeletal abnormalities. Interestingly, Wormian bones are a common feature in CCD, which have been proposed to stabilize the skull anatomy in cases of delayed suture closure. 38
The bony changes underlying CCD are due to autosomal dominant pathogenic variants of RUNX2. RUNX2 is a transcription factor critical for osteoblast progenitor proliferation and differentiation in both intramembranous and endochondral ossification.39,40 It is considered a master organizer of bone formation, which through cooperative binding to DNA sequences regulates the transcription of a variety of osteogenic genes. 41 The pathogenic variants identified in patients with CCD are most commonly located in the Runt domain of the gene, which abolishes the transactivation potential of the protein resulting in functional haploinsufficiency. 42 As a result, complexes of RUNX2 with other transcriptional co-activators required for cell differentiation cannot efficiently bind to osteogenic target genes, as RUNX2 on its own is a weak transcriptional regulator. 43
For patients with CCD, treatment is largely symptomatic as a team of craniofacial specialists is required to address multiple head and neck implications. A primary challenge is dental crowding, which requires orthodontic interventions. 44 If expansion of the cranial sutures is severe, a protective device or helmet may be required. 37 Normally, the anterior fontanelle closes around 10–24 months of age; in CCD, this closure is delayed, sometimes resulting in a persistent anterior fontanelle into adulthood.6,45 Some studies warn clinicians to weigh risks and benefits of repair of bone defects in individuals with CCD due to compromised RUNX2 activity. 46 However, cranioplasty techniques using autologous and alloplastic materials have been successful and non-union in the axial skeleton has been treated with vascularized free fibula grafts.47,48 Older patients are monitored and commonly administered preventative treatment for osteoporosis. Better understanding of how pathogenic variants of RUNX2 affect osteogenic capacity is required to develop precision treatments and assess their compatibility in the clinic.
Extracellular Matrix Production–Yunis-Varón Syndrome
Yunis-Varón syndrome (YVS) is a multisystem disorder with characteristic persistent, large fontanelles, hypoplastic clavicles, a characteristic facial appearance, digit abnormalities, and brain malformations resulting from an organelle-level (lysosome) disruption leading to abnormal extracellular matrix production. 49 Abnormalities of the central nervous system or neurological issues can result in feeding and respiratory difficulties in infants with this condition. 50 In some cases, patients with YVS have life-threatening complications.
YVS is caused by bi-allelic pathogenic variants in FIG4 or VAC14.
51
FIG4 encodes for a phosphoinositide phosphatase and its loss compromises formation of PI(3,5)P252,53; VAC14 encodes an adaptor protein which interacts with FIG4
YVS is an ultra-rare disorder and follows an autosomal recessive inheritance pattern; only about 25 cases have been described since 1980. 21 Most individuals with YVS have a shortened lifespan due to respiratory insufficiency, swallowing, and feeding difficulties. It is difficult to inform future patient management based on such a limited number of clinical descriptions. However, a promising recent study has indicated that chloroquine can correct the enlarged lysosome phenotype in mice, at least temporarily. 59 It is currently unknown whether this treatment translates to clinical use.
Mineralization–Hypophosphatasia
Hypophosphatasia (HPP) is a well-characterised autosomal recessive or autosomal dominant inborn error of metabolism that results in under-mineralized bones and teeth. 60 In severe forms of the condition, the bones are weak and soft resulting in skeletal dysplasia and life-threatening complications. 61 HPP is caused by loss-of-function pathogenic variants in the ALPL gene. 62 ALPL encodes for tissue non-specific alkaline phosphatase (TNAP), an enzyme that hydrolyzes inorganic pyrophosphate, pyridoxal-5’-phosphate, and phosphoethanolamine (PEA). 63 Loss of this enzymatic activity leads to elevated levels of these substrates. Inorganic pyrophosphate inhibits mineralization and is produced by chondrocytes and osteoblasts.64,65 Characteristics of the condition are low levels of alkaline phosphatase in serum and high plasma and urine levels of PEA. 66 The mechanism proposed for HPP involves impaired TNAP function inside bone progenitor cells, involved in mitochondrial respiration and ATP production. 67 For this reason, therapeutic replacement of TNAP enzyme does not alter incidence of craniosynostosis despite improvement of skeletal mineralization, motor, and cognitive function. 68
Severe forms of HPP are estimated to occur at a frequency of 1 in 100, 000. 69 HPP has several phenotypes with varying severity: neonatal, infantile, late, and latent. 70 While all forms of show evidence of defective mineralization, early infantile hypophosphatasia has the appearance of widened sutures due to large patches of unmineralized skull 66 ; this neonatal form is lethal and is associated with multiple fractures. In a cohort study of 25 patients by Kozlowski et al., all subjects had widened cranial sutures. 71 Functional craniosynostosis occurs in these hypomineralized regions, causing a characteristic bulging anterior fontanelle. Precise diagnosis is especially critical in rare bone disorders to guide medical management. 64 For example, high intracranial pressures in the HPP skull indicate a need for skull expansion, in contrast to frequent cranioplasty in CCD to reconstruct skull defects.48,72,73 Improper management due to incorrect diagnosis can thus induce significant harm to the patient. 74
Bone Resorption–Pycnodysostosis
Pycnodysostosis is an autosomal recessive skeletal dysplasia with craniofacial and skeletal features including skull deformities, delayed suture closure, osteosclerosis, acro-osteolysis and bone fragility. 75 In infants, pycnodysostosis manifests as open fontanelles and sutures, frontal and parietal bossing, and maxillary and mandibular hypoplasia. 76 Open cranial sutures have also been observed to persist into adulthood.14,77 Osteosclerosis - abnormal hardening of the bone - advances with growth in pediatric patients and bears increased risk of fracture due to the bone's brittle nature. 78 With approximately 200 patients described in the literature, this rare disorder has a predicted prevalence of about 1.7 cases per million and can necessitate surgical management of craniofacial hypoplasia. 79
Individuals with pycnodysostosis have higher bone mineral density due to loss-of-function pathogenic variants in the CTSK gene, which encodes for Cathepsin K. Cathepsin K is a collagenase required for osteoclastic bone resorption. 80 It is synthesized as a pro-enzyme and is activated by lysosomal cleavage. 81 Quantitative computed tomography (CT) evaluation of bone mineral density in patients with pycnodysostosis shows increased bone volume, which can be explained by the impaired ability of osteoclasts to break down type I collagen matrix. 82 The high susceptibility to fractures may also be due to reduced bone remodeling. However, osteomalacia can also be observed in patients with CTSK variants, suggesting an additional role in bone mineralization. 83 Cathepsin K in osteocytes is likely to play an important role in the suture biology underlying pycnodysostosis and has been hypothesized to be central in mechanosensation and communication with osteoblasts to activate osteogenesis.84,85 The loss of Cathepsin K could influence bone formation and remodeling on several levels, beyond what is discussed in the literature.
Discussion
This literature review presents examples of clinical widening of cranial sutures and fontanelles contextualized within key elements of suture biology, illustrating the variety of causes for similar suture malformations. Cranial sutures exemplify a dynamic continuum between bone formation and resorption. Disruption of the genetic drivers of cell differentiation, extracellular matrix production, mineralization, bone resorption, or any combination can manifest as dramatic craniofacial changes. 86 Suture malformations demonstrate how specific cellular changes can lead to major physiological consequences.
It is unsurprising that the outcome of widened sutures or a persistent fontanelle can arise in multiple ways, given the range of factors governing normal suture development and function, as well as bone formation as a whole. A 1992 case study of an 11-month-old boy with undiagnosed hypophosphatasia further underscores the concept of careful differential diagnosis. The patient was given a presumptive diagnosis of rickets, due to his widened fontanelle, and was treated with high doses of calcium and vitamin D supplementation as a result. However, eventual lab testing revealed reduced ALP levels, hypercalcemia, hypercalciuria, low parathyroid hormone (PTH), and normal levels of vitamin D, which were suggestive of HPP instead of the previously diagnosed rickets. 74 While both rickets and HPP are disorders of bone mineralization, in this case severe clinical complications resulted from indiscriminate treatment.
It is critical to consider the molecular mechanisms underpinning a disorder. Some treatments may not be effective due to mutations in critical pathways. How do the genetic changes underlying the suture malformation interact with the selected treatment modality? This is a crucial element of precision therapeutics, in which clinicians’ evolving ability to ascertain genetic or physiological characteristics of patients can streamline treatment of complex conditions. For example, stimulation of bone formation with a growth factor is dependent on the osteoblast's ability to respond to the factor. 87 Disruption of a critical process like cellular metabolism, as in HPP, impacts other structures in addition to the suture phenotypes described herein. 88 An understanding of underlying bone biology, including gene regulatory networks facilitating normal development, can benefit clinical treatment of rare bone disorders, furthering the field of personalized medicine by carefully considering treatment modality and mechanism in the context of the disordered system. This attention to biological detail can help clinicians to identify the most effective treatments, avoiding those that could cause inadvertent harm.
Study of conditions in which there are abnormal suture outcomes has furthered our understanding of the molecular biology of normal suture development; this is the basis of animal modeling of craniofacial anomalies. In addition to its direct benefit to patients, the study of rare disease biology can also help inform treatments for more common conditions. 8 Thus, there is value in studying rare conditions such as those presented in this review. Indeed, the literature describes several bone disorders presenting with widened cranial sutures with undetermined mechanisms89–94 In addition to the direct effects of gene disruption on osteogenesis or mineralization, ubiquitous facets of cell biology such as epigenetics or mechanotransduction also influence the phenotype.95,96 An example is Craniostenosis, Deafness, Anal abnormalities, and Genitourinary malformations with Skin rash (CDAGS) syndrome, in which disruption of nuclear RNA and intron splicing due to a rare biallelic variant in the RNU12 gene causes a suite of multi-system malformations, including delayed closure of cranial sutures. 97 While the underlying spliceosomopathy has been identified, the downstream effect of minor spliceosome alteration on bone formation leading to delays in CDAGS syndrome is still unknown. The mechanism by which metopism, a persistent metopic suture, occurs is similarly unclear. Genetic underpinnings are the most likely explanation, especially in the context of evolution.98,99
Biology is rarely linear, and these disorders are likely the result of compounding effects of the gene variant or variants on several interacting gene regulatory networks or biological processes. Hence, the identification of the gene affected by pathogenic variation may not be sufficient to fully understand the underpinnings of disordered bone.
The four cases presented in this narrative review illustrate concepts of progenitor cell differentiation, bone mineralization and resorption, and extracellular matrix production; however, these processes do not exist in isolation. The interconnected nature of bone biology dictates additional axes of involvement in craniofacial suture widening. Cleidocranial dysplasia is directly connected to RUNX2 pathogenic variants clinically, but little exploration of the interplay of abnormal RUNX2 with other processes and gene regulatory networks is evidenced in the clinical literature. In contrast, foundational research on Runx2 and understanding of this master bone regulatory gene in non-clinical contexts is vast. 100 Epigenetic regulation of Runx2 through transcriptional silencing, poising cells for expression, and induction of osteoblast differentiation is intricately connected at many levels, which should be considered in the context of the processes and disorders discussed in this review. 101 Other overarching processes governing cell-cell interaction and response to the environment, such as mechanotransduction or lineage commitment, may influence the phenotype or severity of the effects of pathogenic variants. 102 Nevertheless, it is clear from in vitro studies replicating the in vivo suture microenvironment that countless unidentified factors and forces influence bone differentiation.103,104
When a clinician is presented with a patient with widened sutures or fontanelles with delayed closure, it warrants a thorough pediatrics assessment. The mean age of closure for the anterior fontanelle is approximately 14 months of age. By 2 years of age the anterior fontanelle is closed in 96% of children. 105 Non-genetic causes of a large fontanelle include hydrocephalus, hypothyroidism, and rickets. It is important not to miss these treatable conditions. If these conditions have been ruled out and the fontanelle is open after 2 years of age, consideration should be given to an evaluation by a medical geneticist. Red flags that warrant medical genetics consultation, including early referral, are dysmorphic features, developmental delay, congenital anomalies, or additional skeletal anomalies such as unusual body proportions. Genetic testing may involve chromosomal microarray, single gene testing, gene panel testing or exome sequencing depending on the findings of the assessment. The results of genetic testing not only aid the diagnosis of rare diseases but also help inform recurrence risks for future pregnancies. The challenge with rare genetic disease is that there are very few cases and there are rarely established medical management guidelines. Frequently, the natural history of these conditions is not well established. Treatment and surveillance are typically tailored to the individual patient. We highlighted several conditions in this narrative review. The only condition we reviewed for which a targeted therapeutic intervention exists is hypophosphatasia. The therapeutic intervention consists of an enzyme replacement therapy for the infantile and juvenile types, we should be initiated early for best outcomes. Cleidocranial dysostosis, Yunis-Varón syndrome, and pycnodysostosis do not currently have medical management guidelines. While the enlarged fontanelle may eventually close over time in these conditions, there are individuals with cleidocranial dysostosis whose fontanelle remains open throughout life. Individuals with these conditions are managed in a multidisciplinary team with a considered approach to surgical intervention when appropriate. Identification and proactive consideration of underpinning genetic mechanisms should inform these personalized, interdisciplinary treatment plans whenever possible.
The future of bench-to-bedside translation of knowledge and clinical intervention options for patients affected by cranial dysmorphology is teeming with potential. The combination of foundational principles observed and capitulated in pre-clinical animal models with known clinical phenotypes and outcomes from affected patient populations will ignite novel avenues of diagnostic and therapeutic exploration. Drawing from recent successes in preclinical animal models of craniosynostosis, similar efforts may be possible to drive homeostatic maintenance of cranial sutures which fail to grow properly. 106 Future pathophysiological studies will provide a better understanding of suture maintenance, stem cell biology, and osteogenic differentiation cues in the context of pathologically widened cranial sutures. Importantly, the convergence of clinical, basic science, and medical engineering experts is crucial to the development of disease mechanism-based therapeutics to help translate improved treatment to patients with cranial suture pathologies.
Conclusions
Widening of cranial sutures is a rare craniofacial outcome revealing what is not yet known about the causes and implications of cranial suture malformation. The spectrum of craniofacial disorders involving widened sutures described here is caused by pathogenic variants in genes required for the cellular processes governing suture formation and function. Each of the cases presented in this review shares a common feature: widened cranial sutures, or a persistent anterior fontanelle. However, each pathology arises from a different underlying biological perturbation. Whereas distinctions between causes of suture widening are difficult to make clinically, a wholistic view of the disorder and potential biologically relevant causes is required for effective personalized treatment plans for individual patients. Importantly, widened cranial sutures or persistent fontanelles are not simply the inverse of craniosynostosis. This paradigm emphasizes consideration of the underlying biology as patient-specific interventions evolve. This narrative review has illustrated significant gaps in understanding of complex craniofacial disorders in the context of pathologically widened cranial sutures, highlighting avenues for exciting future work to unite foundational biology with clinical practice.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Alberta Dental Association and College Chair in Oral Health Research