
Abstract
Objective
Describe the intelligence quotient (IQ) of children with Pierre Robin sequence (PRS).
Design
Prospective cohort study.
Setting
Neurodevelopmental follow-up clinic within a hospital.
Patients
Children with PRS (n = 45) who had been in the Neonatal Intensive Care Unit (NICU) were classified by a geneticist into 3 subgroups of isolated PRS (n = 20), PRS-plus additional medical features (n = 8), and syndromic PRS (n = 17) based on medical record review and genetic testing.
Main Outcome Measure
Children with PRS completed IQ testing at 5 or 8 years of age with the Wechsler Preschool and Primary Scale of Intelligence, Third Edition (WPPSI-III) or Fourth Edition (WPPSI-IV) or the Wechsler Intelligence Scale for Children, Fourth Edition (WISC-IV) or Fifth Edition (WISC-V).
Results
IQ scores were more than 1 to 2 standard deviations below the mean for 36% of the overall sample, which was significantly greater compared to test norms (binomial test P = .001). There was a significant association between PRS subtype and IQ (Fisher’s exact P = .026). While only 20% of children with isolated PRS were within 1 standard deviation below average and 35% of children with syndromic PRS were below 1 to 2 standard deviations, 75% of PRS-plus children scored lower than 1 to 2 standard deviations below the mean.
Conclusion
PRS subgroups can help identify children at risk for cognitive delay. The majority of children with PRS-plus had low intellectual functioning, in contrast to the third of children with syndromic PRS who had low IQ and the majority of children with isolated PRS who had average or higher IQ.
Pierre Robin sequence (PRS), occurring in ∼1:8500 to 14 000 children, 1 is characterized by micrognathia, glossoptosis, and airway obstruction (UAO). 2 Cleft palate is commonly associated. 2 It is often first recognized in newborns with potentially life-threatening breathing and feeding difficulties and failure to thrive. 3 PRS is phenotypically heterogeneous with a spectrum of severity and uncertain underlying pathogenesis. 4 Some children with PRS present with traditional PRS features associated with a known syndrome (“Syndromic PRS”); the 2 most common being Stickler syndrome (SS) and chromosome 22q11 deletion syndrome. 1 A wide range of chromosomal abnormalities, skeletal dysplasias, neuromuscular disorders, and teratogenic exposure may be present. 1 In contrast, children with non-syndromic PRS present either with only the core features (“Isolated PRS”), or with additional malformations inconsistent with a known syndrome or genetic condition (“PRS-plus”).1,4 The phenotypic spectrum of PRS-plus is broad, with craniofacial dysmorphology (eg, hypertelorism, low-set ears, and preauricular skin tags), muscular skeletal anomalies (eg, congenital dislocated hips, scoliosis), and ocular anomalies (eg, strabismus) commonly observed. 4
Literature suggests that congenital abnormalities and medical complications associated with PRS place children with this condition at increased risk of poor neurodevelopmental outcomes. 1 Brainstem dysfunction may exist in the prenatal and neonatal periods, which may have implications for later neurodevelopment. 5 Feeding and respiratory complications in infancy and childhood, such as obstructive apnea and failure to thrive, have the potential to disrupt the nervous system and impact neurodevelopment.1,2 Hearing loss is a common comorbidity, which has implications for language development and learning abilities.6,7 However, evaluation of the neurodevelopmental outcomes of children with PRS remains scarce and further investigation is warranted.
Preliminary research evaluating cognitive outcomes focused on isolated PRS, with 1 study also including SS.8,9 Despite varied methodologies across these studies, the majority of children with isolated PRS and SS demonstrated age-appropriate cognitive function.8,9 Nonetheless, both groups showed mildly reduced cognition compared to normative data 8 and healthy controls. 9 Furthermore, Thouvenin et al. 8 reported severely impaired cognition in 8% of children with isolated PRS, concluding that PRS is associated with a greater risk of severe cognitive impairment relative to the general population. 8 Differences in cognitive outcomes between isolated PRS and SS were evident but not significant, with relatively lower functioning observed in SS. 8 The neurodevelopmental outcomes of the broader PRS phenotype were not investigated in these studies.
This study aimed to report on the longer-term overall intellectual outcomes of children with isolated PRS, PRS-plus, and syndromic PRS. We primarily aimed to document and compare the overall intelligence of children with PRS against normative data, and to compare the overall intelligence of the different PRS subtypes. Given the increased risk of language difficulties, our secondary aim was to explore the verbal intellectual abilities of these children.
Methods
Participants
Ninety infants with PRS presented to the Neonatal Intensive Care Unit (NICU) at The Royal Children's Hospital (RCH), Melbourne, Australia from January 1, 2004 to June 30, 2014, of whom 5 died aged between 6 weeks and to 3 years of age. The neurodevelopmental outcomes of these children were prospectively followed in our dedicated Neonatal Neurodevelopmental Follow-up Clinic and included 20 children with isolated PRS, 8 with PRS-plus, and 17 with syndromic. Only Victorian residents were offered neurodevelopmental surveillance at 5- and/or 8 years old in our clinic. This surveillance involved neuropsychological assessment. There were no data available to us regarding eligible children who did not attend for clinical follow-up. The review age was predetermined for all children on the basis of anticipated clinical need and benefit, with consideration of critical periods of development and academic transition. The RCH Human Research Ethics Committee approved clinical data use for research with an “opt-out” option from 2015. Prior to this, caregivers provided written consent for data to be used for research.
Measures
Data were collected on paper records and stored in Epidata. 10 The assessing clinician obtained data on PRS characteristics and variables that may influence neurodevelopmental outcomes including gestational age, birthweight, Apgar scores, sex, tube feeding, mandibular distraction osteogenesis (MDO), tracheostomy in the NICU period, and the presence of neuroimaging abnormality from electronic medical records. Hearing impairment data was collected from parents when this was not available on file. Children were classified into the 3 PRS subgroups based on medical record review of clinical findings and genetic testing results (assessed by TYT and SSWC). Single-nucleotide polymorphism (SNP) microarray is routinely performed at RCH in all patients with PRS. Parents indicated the level of maternal education (lower: ≤ 12 years, higher: >12 years) and the primary language spoken at home (English or Other). The Index of Relative Social Advantage and Disadvantage (IRSAD) was employed as a proxy measure of socioeconomic status. This is based on the 2011 Australian census data for the participants’ postcodes at birth. 11 IRSAD scores were analyzed as deciles with lower deciles reflecting greater disadvantage.
Children underwent clinical neuropsychology assessment using one of the following validated, age-standardized, and gold-standard intelligence measures: the Wechsler Preschool and Primary Scale of Intelligence, Third Edition (WPPSI-III) 12 or Fourth Edition (WPPSI-IV), 13 or the Wechsler Intelligence Scale for Children, Fourth Edition (WISC-IV) 14 or Fifth Edition (WISC-V). 15 The Australian Edition of these measures was used, which includes local norms. The measure was selected based on the child’s age and the newest edition available at the time. For children who underwent serial assessment, the results of their most recent assessment are reported. Overall intelligence was estimated by the Full-Scale Intelligence Quotient (FSIQ; M = 100, SD = 15). Considering the high risk of language difficulties, the verbal subdomain of intelligence was also referred to, ie, the Verbal Comprehension Index (VCI)/Verbal IQ (VIQ; M = 100, SD = 15). Other subdomains were not considered for current analyses due to inherent variation in procedures between different editions of the tests. Scores were converted to z-scores (M = 0, SD = 1) and treated categorically. Scores considered within or above age expectations were defined as follows: within age expectations or age appropriate z = −1 to < + 1 (ie, within 1 SD of the mean), above age expectations z = + 1 to < + 2 SD (ie, between 1 and 2 SD above the mean), and well above age expectations z = > + 2 SD (ie, 2 SD or more above the mean). Scores considered within the impaired range were defined as follows: mild impairment or mildly below age expectations z = −2 to < −1 (ie, between 1 to 2 SD below the mean); and moderate to severe impairment or well below age expectations z = < −2 (ie, 2 SD or more below the mean). Comparison is made to the normative sample distribution of scores provided with the test.
Statistical Analyses
Statistical analyses were computed in Stata 14. 16 Descriptive statistics were reported using means and standard deviations for continuous variables, or medians and interquartile ranges for nonparametric variables. Categorical data were described using counts and percentages. The proportion of children in the combined sample (ie, with any type of PRS) with any impaired range score was compared to test norms using a binomial test. Given the small sample size, Fisher’s exact test (3 × 5) analyzed the proportion of children in each PRS subgroup with FSIQ scores falling within the average versus impaired ranges. Similarly, the proportion of children in each PRS subgroup with VCI/VIQ scores falling within the average versus impaired ranges was analyzed using Fisher’s exact test.
Results
Outcomes of children with isolated PRS, PRS-plus, or syndromic PRS are reported. Clinical neurodevelopmental surveillance at 5- and/or 8 years old was offered to 77 children. Attendance was declined for 16 eligible children and 14 children could not be contacted. The remaining 47 attended for cognitive assessment, with complete data obtained for 45 of these children (19 5-year olds and 26 8-year olds). Overall FSIQ and VCI/VIQ scores could not be calculated for 2 children with incomplete intelligence data due to task refusal. All participating children primarily spoke English at their most recent assessment.
Children were classified as follows: isolated PRS n = 20, PRS-plus n = 8, and syndromic PRS n = 17. The syndromic PRS subgroup comprised children with the following syndromes: SS (n = 9), Catel–Manzke syndrome (n = 1), chromosome 22q11 deletion syndrome (n = 1), Duane syndrome (n = 1), chromosome 22q11.2 Duplication syndrome (n = 1), Hajdu–Cheney syndrome (n = 1), congenital disorder of glycosylation type 1t (n = 1), Rapp–Hodgkin syndrome (n = 1), and Smith–Magenis syndrome (n = 1). The kinds of additional malformations in the PRS-plus group varied and were idiosyncratic. Examples include cardiac defects (eg, ventricular septal defect), renal defects (eg, duplex or horseshoe kidney), neuroanatomical malformations (eg, Chiari malformation, agenesis of the corpus callosum, and cerebellar atrophy; n = 3), microcephaly, and other facial abnormalities (eg, left facial nerve palsy and hypoplasia, bulbous nasal tip, and curved eyebrows).
Table 1 shows the demographic and clinical characteristics of all children who participated in the assessment. Children with each PRS subtype were typically born at term with a normal birthweight and a healthy Apgar score at 5 min (ie, scores >7). Over half of the children with PRS required tube feeding and MDO surgery. No children underwent tracheostomy during their NICU admission. Neuroimaging was performed in 39 children before 2 months old (10 MRI and 38 cranial ultrasound), with abnormalities evident in 21% of all children with PRS, none of which were in the isolated PRS group.
Demographic and Clinical Characteristics of Children With PRS who Underwent Intellectual Assessment.
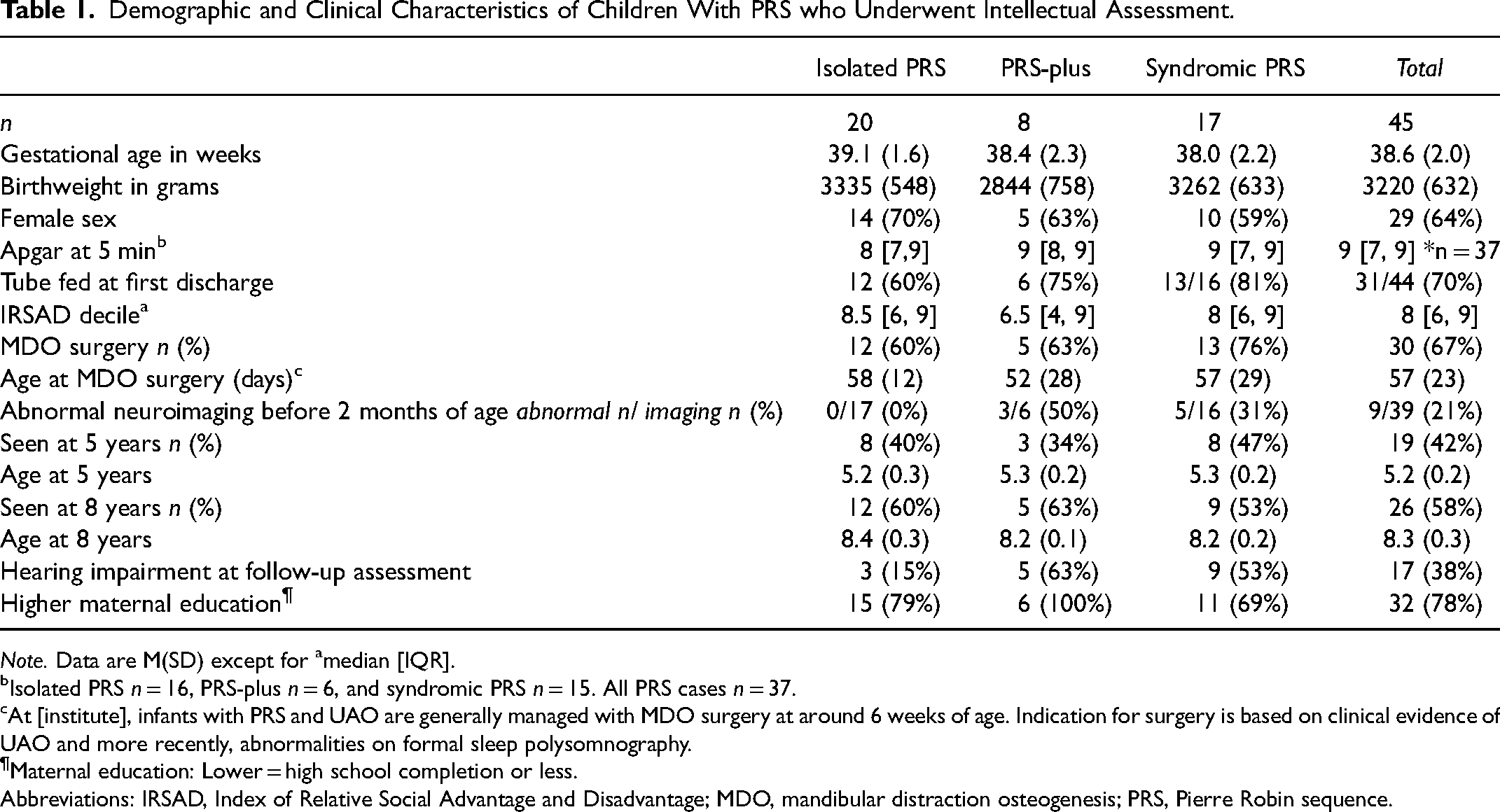
Note. Data are M(SD) except for amedian [IQR].
Isolated PRS n = 16, PRS-plus n = 6, and syndromic PRS n = 15. All PRS cases n = 37.
At [institute], infants with PRS and UAO are generally managed with MDO surgery at around 6 weeks of age. Indication for surgery is based on clinical evidence of UAO and more recently, abnormalities on formal sleep polysomnography.
Maternal education: Lower = high school completion or less.
Abbreviations: IRSAD, Index of Relative Social Advantage and Disadvantage; MDO, mandibular distraction osteogenesis; PRS, Pierre Robin sequence.
Table 2 shows the mean, median, and range of intelligence quotient (IQ) scores for the PRS subgroups and entire sample. Figure 1 shows categorical IQ scores for the combined sample and for each PRS subgroup, as well as the normative distribution of scores. In the combined sample, over a third (36%) of children with PRS scored > 1 SD below the M on FSIQ. This was significantly greater than expected compared to test norms (binomial test P = .001). Of all children with PRS, 18% scored > 2 SD below the M, compared to only 2% of the normative population.
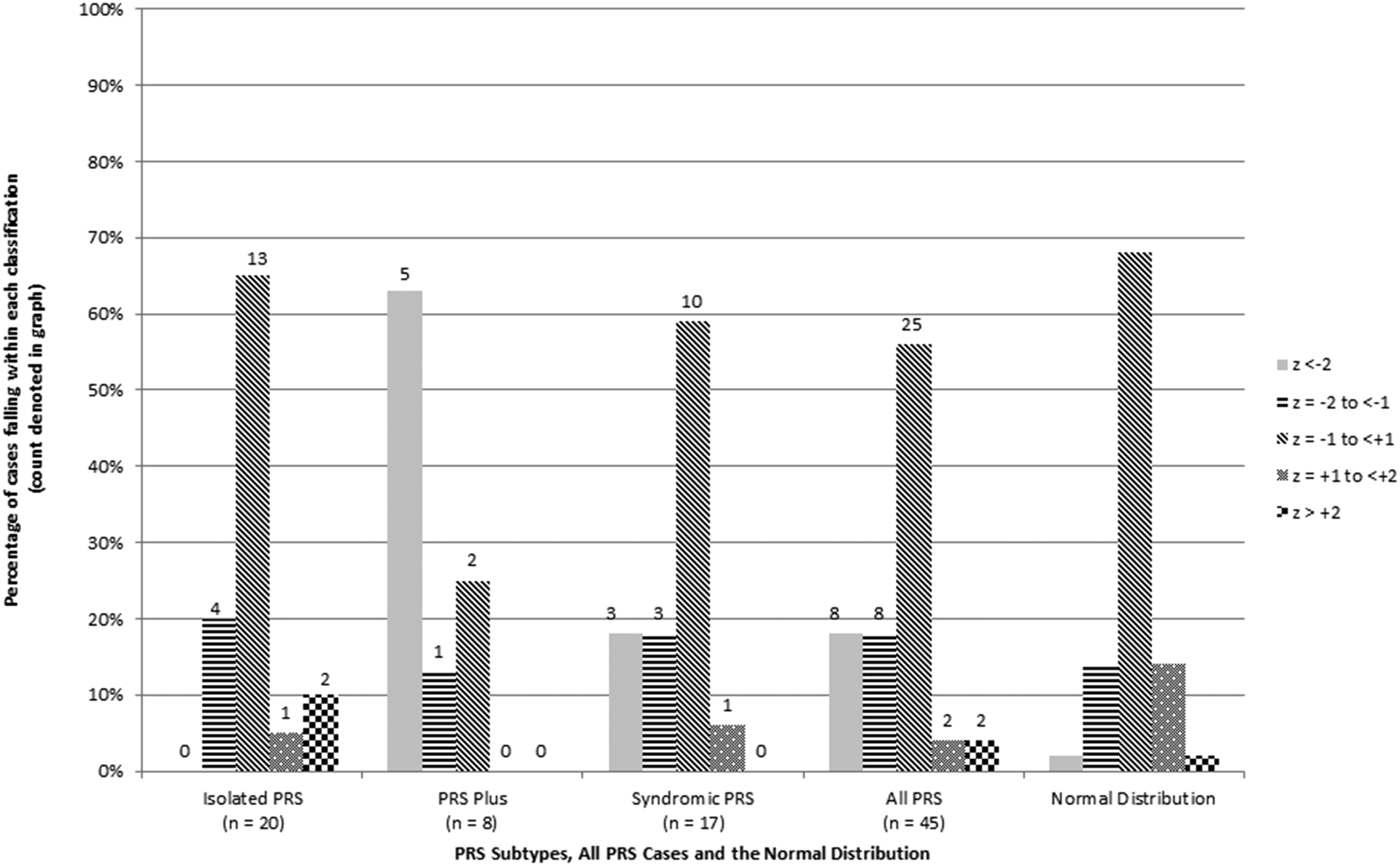
Shows categorical IQ scores for the combined sample and for each PRS subgroup, as well as the normative distribution of scores.
Mean, Median, and Range of Intelligence Quotient (IQ) Scores.

Note. Data are missing for a child in the PRS-plus subgroup. This child’s score was known to fall >2 SD below the mean, but the exact value was not available.
There was a significant association between PRS subtype and FSIQ (Fisher’s exact P = .026). The majority of children with PRS-plus scored in the impaired range on FSIQ (ie, 75% showed mild to severe impairments) and no children displayed above average FSIQ. The syndromic PRS subgroup exhibited greater variability in their outcomes, with 35% showing mild or greater impairments in FSIQ and the remaining 65% of children performing at or above age expectations. While the isolated PRS group contained some children within 1 standard deviation below the mean (20%), the majority of these children demonstrated FSIQs within or above age expectations. Therefore, relative to the isolated PRS group, PRS-plus, and syndromic PRS subgroups deviated more from the normative population in terms of rates of impairments versus expected or above expected scores.
Figure 2 shows VCI/VIQ of the combined sample and each PRS subgroup. The distribution of VCI/VIQ scores appeared comparable to those of the FSIQ scores for each subgroup. However, there was a significant association between PRS subtype and VCI/VIQ (Fisher’s exact P = .002). The majority of children with PRS-plus displayed moderately to severely impaired VCI/VIQ, while scores in the other subgroups were more consistent with the normal distribution.
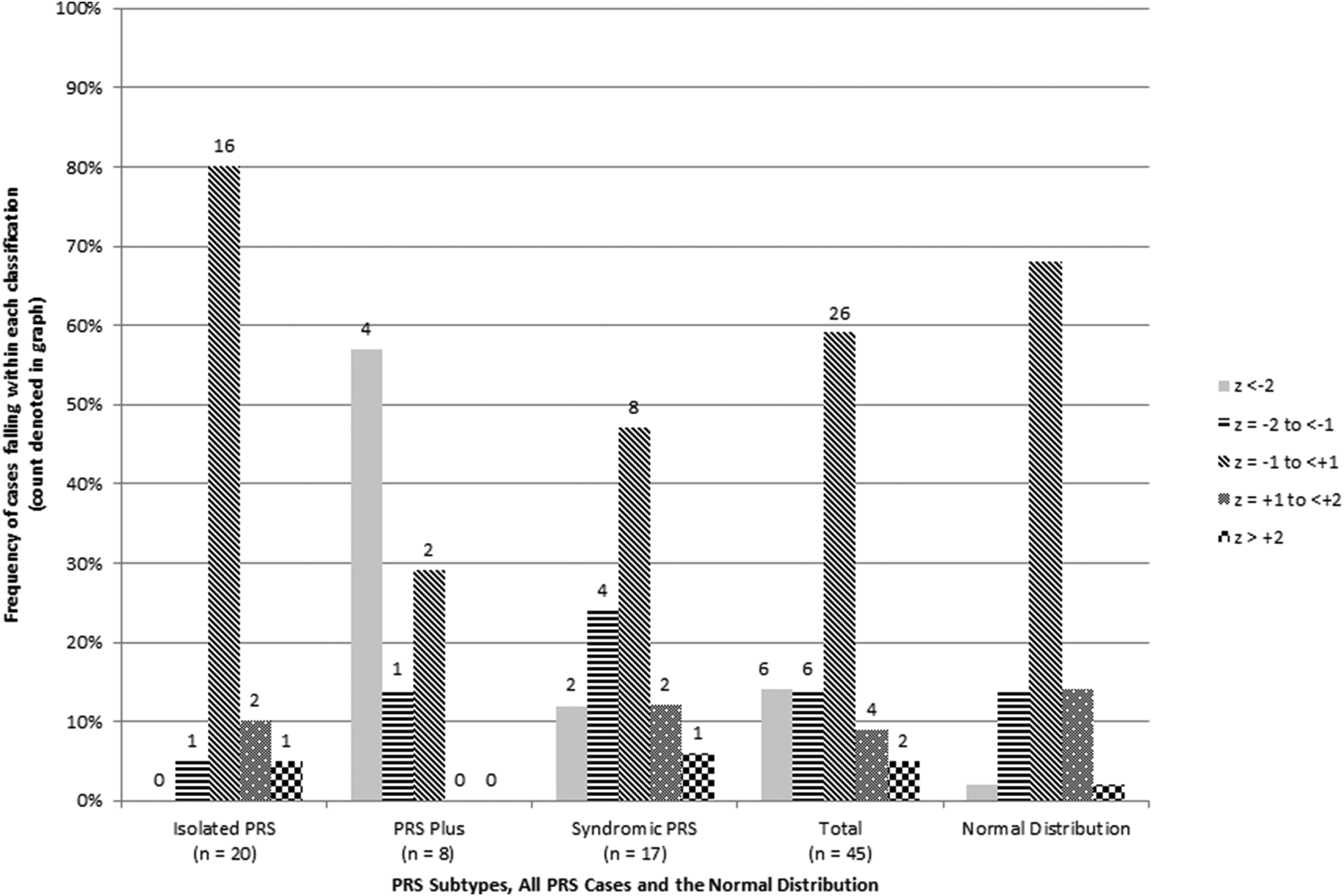
Shows VCI/VIQ of the combined sample and each PRS subgroup.
Discussion
Our findings show children with PRS are more vulnerable to cognitive problems, with over a third of our overall sample displaying some level of intellectual difficulty at 5- or 8 years old. This is more than double that expected with reference to the normative data. The prevalence of moderate to severe impairments (ie, FSIQ scores placed below 70) was also much higher in our overall sample (ie, 18%) as compared to the rates within the normative data (∼2%). Our finding of higher rates of intellectual impairment in children with PRS relative to the general population is consistent with previous research. 8 However, subgroup level analyses in this study revealed substantial differences in the frequency of intellectual impairment across the PRS subtypes.
Most children with isolated PRS performed within or above age expectations, with only 20% showing mild difficulties and none placed in the moderate to severely impaired range (ie, none had IQ scores below 70). In contrast, the majority of children with PRS-plus showed mild or more severe impairments, with an overwhelming 63% displaying FSIQ scores below 70. Notably, around one-third of PRS-plus children presented with neuroanatomical malformations that are associated with an increased risk of cognitive impairment.17,18 Children with syndromic PRS were more varied in their outcomes, likely reflecting the diverse syndromes ascertained in our sample. More than half of these children were at or above expectations, yet a larger than expected proportion still scored below expectations compared to population norms. Overall, the phenotypic spectrum and expected genetic heterogeneity associated with PRS 4 likely accounts for the varied intellectual outcomes observed in this study. Indeed, genetics are known to play an important role in the etiology of intellectual impairment, where genetic mutation, deletion, or rearrangement affecting gene encoding and in turn protein function can have severe consequences for brain development and cognitive functioning. 19
Our study reports overall intellectual outcomes of syndromic and non-syndromic PRS broadly, building on the limited body of existing literature in this condition. Drescher and colleagues 9 first evaluated the cognitive outcomes of children with PRS and found age-appropriate performances. However, it is unclear to what extent their sample comprised children with isolated or other forms of PRS. Thouvenin and colleagues 8 evaluated children with isolated PRS and syndromic PRS, particularly SS, for which they found age-appropriate long-term cognitive development. Our finding of largely average or higher intellectual functioning in the isolated PRS group is consistent with this prior literature. In contrast, difficulties in the syndromic PRS group were greater than previously identified due to our inclusion of a broad range of syndromic diagnoses.
Given the elevated prevalence of hearing impairment that places children with PRS at risk of language delays, 8 verbal intellectual functioning was also explored in the present study. Results showed a significant association between PRS subtype and verbal intelligence. Impairment was more frequent in children with PRS-plus and syndromic PRS compared to isolated PRS; however, children with PRS-plus appeared particularly vulnerable to difficulties. Overall, the frequency of verbal intellectual impairment largely mirrored the difficulties observed for overall intelligence, with the exception of somewhat higher rates of typical verbal intelligence compared to typical overall intelligence seen in the isolated PRS group. This finding may reflect the low rate of hearing impairment at the time of assessment and a possible weakness in other intellectual domains for the subset of children with isolated PRS.
Our findings reinforce the importance of screening for cognitive difficulties in children with PRS and a multidisciplinary approach to care. However, we suggest prioritizing children with PRS-plus and syndromic PRS for assessment, especially in resource-limited settings. These children are more vulnerable to intellectual impairment and would benefit from close monitoring and assessment. Neuropsychological assessment provides an understanding of cognitive functioning at critical periods of neurodevelopment, which can, in turn, facilitate the provision of early intervention and inform educational planning, placements, and supports. The involvement of clinical genetic assessment can accurately classify a child’s PRS subtype, assisting other clinicians to identify children who are at greatest risk of intellectual impairment.
Strengths of this study include the reporting of all PRS subtypes and evaluation using a widely administered standard measure of intelligence. This methodology likely explains some of the differences in our findings relative to others in the field. In this context, we have been able to provide key information that has direct implications for clinical care. Our sample was collected only from children with PRS who had been in the NICU and may not represent the range of functioning of children who did not present with symptoms severe enough to require inpatient treatment as infants. Also, this sample was part of standard clinical follow-up, thus it may be limited by some inherent selection bias. It is possible that parents of children attending the clinic were those with greater concerns for their child or of greater social advantage. Notably, the children in our sample with PRS-plus were more socially disadvantaged compared to the other PRS subgroups and this may play at least some role in their cognitive outcomes. However, our small sample size restricted detailed statistical analyses exploring correlates of outcome, including social disadvantage as well as consideration of exposure to surgical intervention and other medical risk factors. Routine polysomnography was not yet available to our patient cohort at the time they were managed in the NICU. This is an important factor for future investigations. Replicating findings in large sample sizes across all subgroups is needed, including allowing for greater distinction among syndromes associated with PRS and inclusion of less severe presentations of PRS. In addition, future studies should look beyond verbal abilities to other aspects of intellectual functioning including nonverbal skills, information processing speed, and working memory. The inclusion of adaptive functioning measures in future research would help clarify diagnostic criteria for low-functioning children. Finally, a history of resolved hearing impairment was difficult to document and restricted to the presence or absence of hearing difficulties at the time of study assessment.
In this study, children with PRS-plus were largely in the range of functioning consistent with intellectual disability and about a third of children with syndromic PRS also appeared to be at increased risk of cognitive difficulties. Children with isolated PRS were generally in the average range or higher, but did have around a fifth of children with some mild delays. Together, these findings suggest different neurodevelopmental trajectories across the PRS subtypes; however, further research is required to understand the broader cognitive profile and risk factors for poor outcomes in each group. Given the relatively low incidence of this condition, development of international registries would facilitate enhanced data collection. We have highlighted the importance of employing genetic assessment for accurate clinical phenotyping in future research. In the clinical setting, genetic testing and classification are crucial to ensure adequate targeted long-term neurodevelopmental follow-up for children at greatest risk, especially those with PRS-plus.
Footnotes
Acknowledgments
We gratefully acknowledge funding from The Royal Children's Hospital Foundation. The funder had no role in the present study.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.