
Abstract
Gene therapy has become a widely accepted treatment for inherited or acquired genetic diseases. Lentiviral vectors are of particular interest because of their favorable biosafety profile and ability to introduce their therapeutic cargo into non-dividing cells. For clinical use, these viral vectors must be generated under conditions of good manufacturing practice in large quantities, which currently are provided via transient production. A solution for stable, robust, easy to scale, cost-effective, and predictable production of the therapeutic vectors is currently not available. Here, we describe the design, generation, and characterization of EL1-820, a packaging cell line for the stable production of lentiviral self-inactivating (SIN) vectors pseudotyped with the envelope glycoprotein of vesicular stomatitis virus. EL1-820 enables the introduction of a lentiviral SIN-vector expression cassette via Flp-recombinase–mediated cassette exchange (RMCE) into a predefined locus selected for optimal vector production, with expression units designed to improve reliability. EL1-820–based producer clones generated similar titers (1 × 107 TU/mL) from a targeted, single-copy integration of a lenti-GFP or a lenti-chimeric antigen receptor transfer vector as transient production. In initial scale-up experiments, multiple harvests from bioreactors could be achieved, resulting in titers of around 8–9 × 107 TU/mL after tangential flow filtration and a total yield of about 2.3 × 1011 TU. In conclusion, RMCE-based introduction of the transfer construct allows stable, defined, predictable, and safe vector production suitable for clinical applications.
Keywords
INTRODUCTION
Gene therapy aims to alter the genetic properties of target cells and is evolving into a routine treatment for inherited and acquired diseases. Its growing usage increases the demand for vectors to deliver the therapy. For over two decades, lentiviral vectors (LVs) have been used to stably modify cells in vitro or in vivo. 1 Even cells difficult to target, like brain or stromal/mesenchymal stem cells, can be transduced successfully.2–4 Many researchers and almost all clinical trials working with LVs use transiently produced vectors, pseudotyped with the envelope glycoprotein of vesicular stomatitis virus (VSV.g), enabling broad cell-type transduction. Both integrating and non-integrating viral vector systems exist; however, for long-term expression of therapeutic transgenes, integrating vectors are preferred.
Despite the clinical potential of gene therapy, major challenges exist to consistently manufacture therapeutic vectors via transient production: Scaling-up production is difficult, and commercial applications might be limited due to increased costs for quality assessment of starting materials and batch release testing. 5
To overcome these constraints, stable producer lines can be generated to manufacture safe, cost-effective, and reproducible vectors. The first stable, efficient lentiviral producer cells used gamma-retroviral vectors (gRVs) for transmission and expression of lentiviral packaging components. 6 These cells generated high LV titers; however, long terminal repeat (LTR)-driven gene expression and only partial sequence optimization led to considerable recombination of replication competent LVs (RCL). Consequently, self-inactivating (SIN) gRVs were developed to transfer packaging components into 293T cells, creating GPRG (gag-pol, rev, and VSV.g) cells. 7 The sequence-optimized packaging components were expressed from internal promoters with or without tet-control. To introduce the lentiviral therapeutic SIN-vector into GPRG cells, a concatemeric DNA-array allowing stable transfection of at least 25 tet-inducible expression cassettes was developed. Despite numerous reports of stable lentiviral packaging cell lines,6,8–20 GPRG cells are, to our knowledge, the only successful LV-producer cell line used in clinical trials.21–23
Stable producer lines offer clear advantages compared to transient production, but the complexity of the lentiviral genome and the partially toxic packaging components make their generation challenging. For example, to prevent RCL formation, regions of sequence homology between crucial viral cis-elements, essential viral genes, and the therapeutic vector need to be eliminated by sequence optimizations. 24
In the past two decades, improvements in codon usage optimization, translational efficiency, structural requirements,25,26 and the balanced ratios of packaging components 10 have advanced stable LV production. Furthermore, improvements in fixed-bed bioreactor technology enabled the usage of adherent producer cells for large-scale vector production, for example, in the Hyperstack or Ascent bioreactors.
We developed a system incorporating multiple improvements for reliable and stable generation of clinical-grade LV with scale-up potential. Our producer cells stably express optimized packaging components and allow site-directed integration of a single customized SIN-vector, enabling consistent LV production in bioreactors.
MATERIALS AND METHODS
Cell culture
293T (ATCC # CRL-11268) cells, their derivatives, and HT1080 (ATCC # CCL-121) cells were cultured in Dulbecco’s modified Eagle medium (DMEM; Invitrogen) supplemented with 10% heat-inactivated fetal bovine serum (FBS; Cytiva) at 5% CO2 and 37°C. EL1-tr/-trg/-trgv and -820 cells were cultured in the presence of 100 ng/mL doxycycline (Sigma-Aldrich) to keep tet-inducible units in the “off-state” during expansion. Cultures were propagated at logarithmic growth and passaged at 70–80% confluency. Cells were harvested after incubation for 2–4 min with TrypLE Select (Invitrogen) and transferred into fresh medium or used for analysis. Jurkat (DSMZ, ACC282) cells were cultivated in Roswell Park Memorial Institute 1640 medium (Gibco) supplemented with 10% FBS at 5% CO2 and 37°C. The used and generated cell lines are summarized in Supplementary Table S2.
Plasmid construction
The gamma-retroviral MT-series of vectors were developed based on the ES.12 SIN-vector. 27 The Ptet-minimal promoters T3i and T6i are based on the Ptet-T3/-T6 minimal promoters 28 , modified to contain intronic-sequences from rabbit beta-globin (T6i) and human CMVie (T3i) within their 5ʹ untranslated regions. Expression plasmids for HIV syn.Rev and HIV syn.Tat were assembled by standard cloning techniques. Both sequences were designed CpG-free to destroy homology to the wild-type sequence, as this sequence is partially present in the viral transfer vector or in the expression constructs of packaging components but is also essential for vector production. After gene synthesis (GenScript), the open reading frames (ORFs) were cloned into pUC57 and then transferred into the respective plasmids.
The wild-type VSV.g envelope glycoprotein was not modified. It was PCR-amplified from pMD.g, subcloned into pBluescript SK II+, sequenced, and subsequently transferred into the MT vector.
To prevent homology-dependent recombination, syn.GP (Fig. 1E) contains an excisable RRE element at its 3ʹ end and was subcloned into pUC57. We have codon-optimized the wt sequence between positions 1–355 (gag) and 3992–4109 (pol) to contain only stretches <8 nt of homologous sequence to design syn.GP. Besides this, codon modifications were done over the residual sequence to destroy splice sites, unwanted restriction sites, or start sites. The ribosomal frameshift region (at position 1296) and the surrounding ±50 nt remained untouched. For testing the efficiency of the different GP-variants (GPwt, syn.GP, or syn.GP4) in transient transfection assays, the respective ORFs were subcloned into the mammalian expression plasmid pcDNA3 by standard cloning techniques. The final expression cassette was subcloned into ES.12.
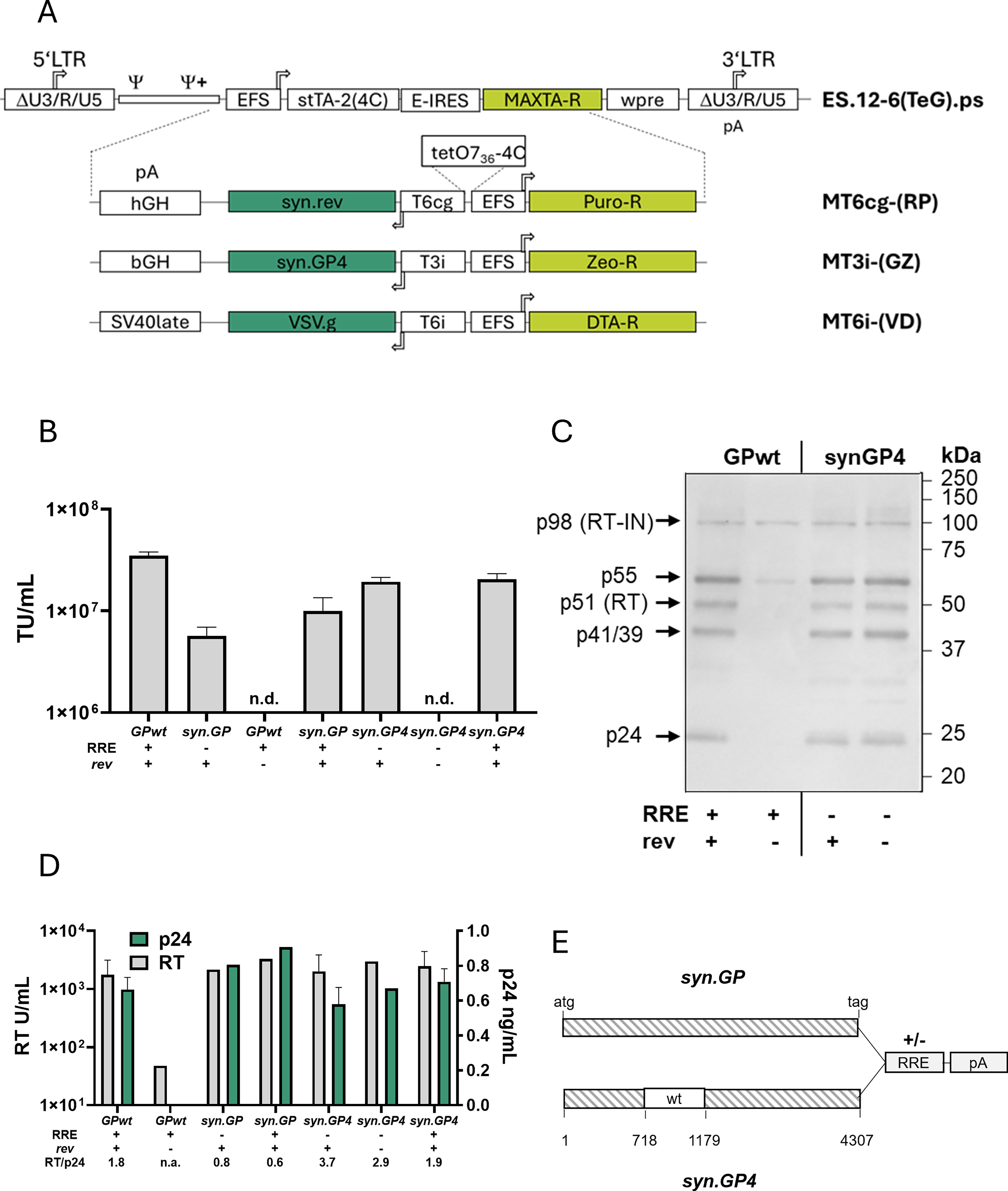
Improved vector design for stable lentiviral packaging cell lines.
The retroviral tagging vector ETAG.fcN has been described previously. 27
Three targeting constructs (Fig. 3B) were cloned based on the pbib-ETAR.fcvi construct described previously. 27 The expression cassette from the lentiviral SIN-vector pRRL.SIN.ppt.GFP.WPRE 29 was inserted into the targeting construct pbib-ETAR.fcvi, resulting in pbib-ELTAR.fc-pRRL (Fig. 3B). Into this backbone, an HIV-syn.Tat-P2A fusion was cloned, interspacing the CMV minimal promoter and the GFP/blasticidin marker gene (GB) and resulting in pbib(t)-ELTAR.fc-pRRL. To generate pbib(tt)-ELTAR.fc-pRRL, the tet-inducible syn.Tat-expression unit was introduced together with a bidirectional inducible promoter (HIVmin/tetO7/pTet-T4 28 ) and the human growth hormone polyadenylation side (HGHpA) for the termination of antisense-directed gene expression. The EFS promoter serves for constitutive expression of the GB-marker gene (Fig. 3B). The putative therapeutic pbib(tt)-ELTAR.fc.CLDN6-CAR vector was generated by replacing the PGK-GFP-WPRE fragment of the LV-RRL vector with an EFS-CLDN6-CAR-WPRE fragment via standard cloning techniques (Fig. 6A).
The LTR-driven lentiviral test vectors (LCO-series) were constructed based on a pRRL.SIN.ppt.GFP.WPRE backbone. 29 The modifications include (1) removal of the internal PGK promoter, (2) exchange of the SIN-U3 region with the RSV-U3 region, and (3) replacing GFP with the ORF of the GB fusion marker gene. For constitutive HIV-tat expression, the syn.Tat ORF was linked to GB via the internal ribosomal entry site of Encephalomyocarditis Virus (E-IRES) (Fig. 2F).
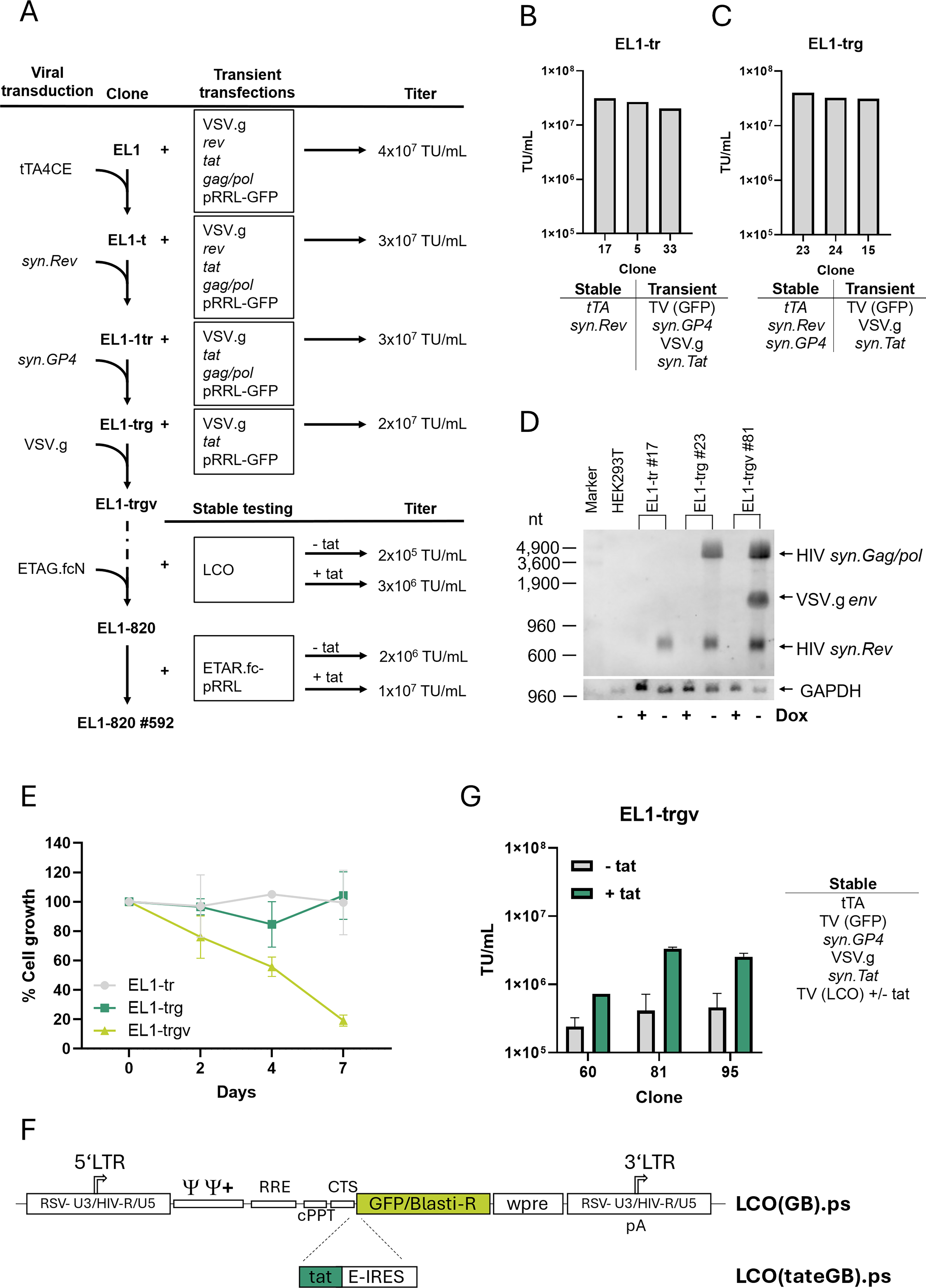
Generation and characterization of EL1-820 packaging cells.
The used and generated plasmids are summarized in Supplementary Table S2.
Testing vector production capacity of intermediate packaging clones
For stable vector production using the EL1-tr/-trg cells, 1 × 104 cells/cm2 were seeded on a six-well dish 3 days prior induction by removing doxycycline. Two days after induction, cells were transfected (q-PEI, Polyplus) with plasmids encoding the remaining genes essential for LV production (Fig. 2A) as previously described. 29 Medium was replaced 16 h after transfection, and 24 h later, viral vector-containing supernatant was harvested, filtered (S06-E750-05-S, Repligen), and stored at −80°C until further usage.
EL1-trgv cells were transduced with LCO-vectors and selected with blasticidin (10 µg/mL, InvivoGen). Approximately 1 × 104 cell/cm2 were plated on a six-well dish for 3 days, and the cells were induced by removing doxycycline. Medium was exchanged every 24 h, collected, filtered (0.45 µm, Cytiva, previously Pall Corporation), and stored at −80°C until further usage.
Stepwise generation of EL1-820 cells
Vectors ES.12-6(TeG).ps (Fig. 1A), the MT-series (Fig. 1A), and ETAG.fcN (Fig. 3A) were produced by three-plasmid transfection in 293T cells as previously described. 27 The respective precursor cell lines were transduced with an MOI of 0.01. Transduced cells were selected with the corresponding drug for 5–7 days (1 µg/mL puromycin; Sigma-Aldrich), 1-fold MAXTA (GPT Selection kit, Merck Millipore), 1 ng/mL DTA (Sigma-Aldrich), 100 µg/mL Hygromycin (InvivoGen), or 100 µg/mL Zeocin (Invitrogen). Approximately 100 clones, ES.12-6(TeG).ps and MT-series or 700 clones (ETAG.fcN) were selected via limiting dilution in 384-well plates (Supplementary Fig. S2), and the resulting EL1-820 clones were then tested for vector production capacity.
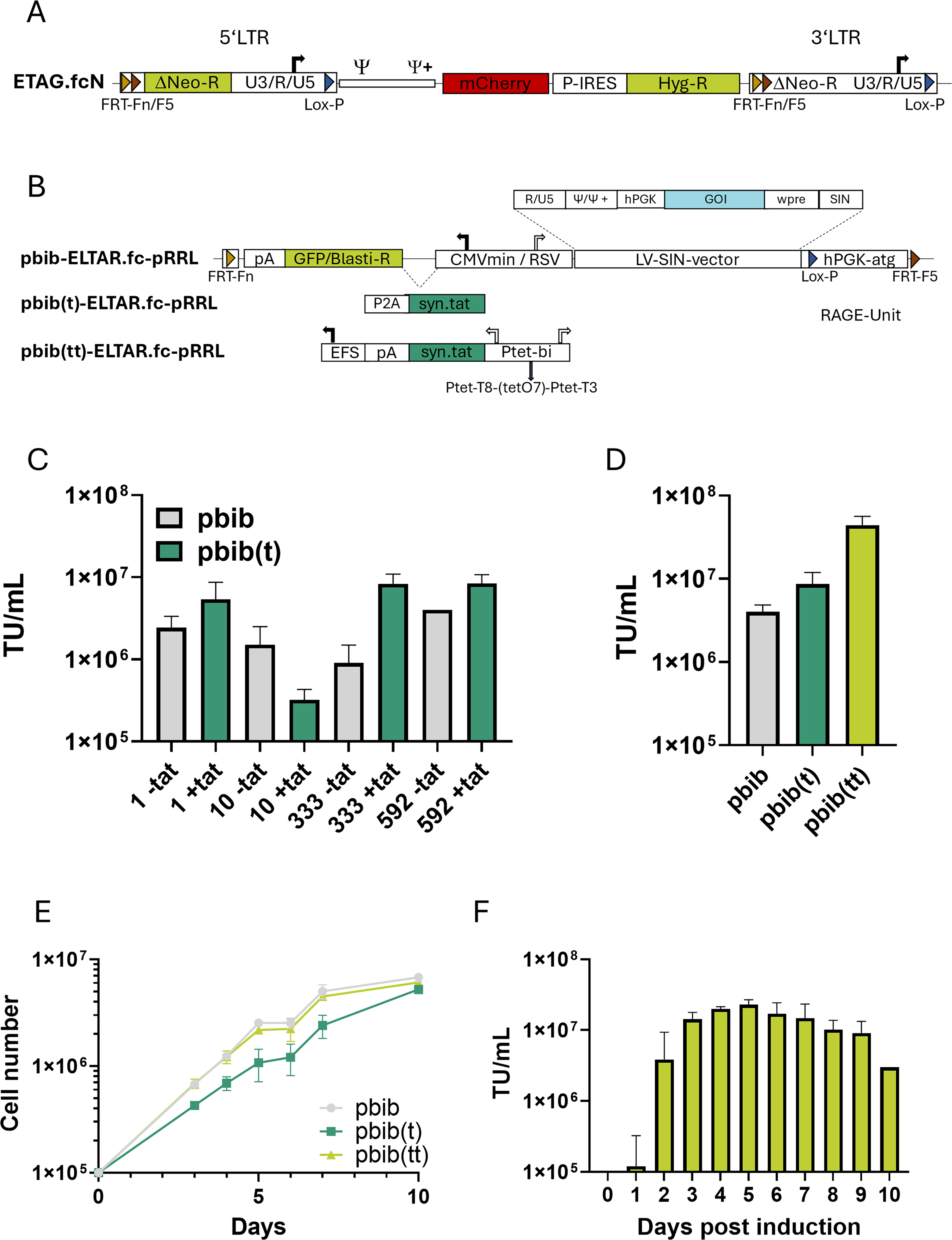
FRT-integration and selection of a tagged packaging cell line.
Integration site analysis via TLA-sequencing was done by Cergentis B.V. (Utrecht, Netherlands).
Titer determination
Titrations of vectors encoding fluorescence proteins (eGFP, GB, or mCherry) were performed on HT1080 cells. Approximately 5 × 105 cells were transduced using a 5-fold serial dilution of vector-containing supernatant with polybrene (8 µg/mL; Sigma-Aldrich) as described. 27 HT1080 cells were harvested 3 days after transduction and analyzed by flow cytometry on a BD Accuri C6 Plus cytometer (BD Biosciences) using BD Accuri C6 Plus software (v1.0). Viral titers were calculated by the relative amount of viable fluorescent (positive) cells relative to non-transduced cells and calculated back to the number of cells initially transduced.
Titers of retroviral vectors for the setup of EL1-820 packaging cells (Fig. 1A) were determined via end-point dilution assays using the respective resistance marker. Approximately 5 × 105 HT1080 cells were transduced with a 10-fold serial dilution of vector-containing supernatants as above. Three days after transduction, HT1080 cells were exposed to the drugs of the corresponding viral vector resistance. The titer was calculated based on the number of resistant colonies after selection for 10–14 days.
Titration of the therapeutic vector expressing the CAR against CLDN6 was performed on Jurkat cells. Briefly, 5 × 105 Jurkat cells were transduced with a 5-fold serial dilution of vector-containing supernatant with polybrene (8 µg/mL). Two days after transduction, cells were harvested, stained with an Alexa Fluor 647-conjugated antibody directed against the Fab portion of the IgG-domain of the CAR protein (goat anti-mouse IgG F(ab′)2 fragment, Jackson ImmunoResearch), and analyzed via flow cytometry. Viral titer was calculated by the ratio of positive to negative viable cells, calculated back to the cells initially transduced.
Targeted cassette exchange and clean-up
For site-specific cassette exchange into EL1-820 clones, 1 × 106 cells were co-transfected (q-PEI) with 0.5 µg Flp recombinase-expressing vector (SV-FLPE 30 ) and 2 µg of targeting construct (pbib-, pbib(t)-, or pbib(tt)-ELTAR.fc-pRRL) encoding the LV. About 18 h post-transfection, cells were transferred to 384-well plates and initially incubated with G418 (Geneticin, Sigma-Aldrich, 1,000 µg/mL) for 10 days, followed by incubation with 400 µg/mL G418 and 10 µg/mL blasticidin (InvivoGen) for another 10 days.
Readthrough of the lentiviral pA signal into the 3ʹ LTR can lead to recombined vectors that can transfer neomycin resistance. This transfer was tested by incubation of about 1 × 106 HT1080 cells transduced with LV-containing supernatant at an MOI of 10. Cells were cultivated for 2 days (to allow amplification of putative neoR-positive clones) before transfer to a larger dish and incubation with G418 (1,000 µg/mL). Colony formation was determined about 10–14 days after the start of selection. 27
To remove unwanted residual tagging vector sequences, in vitro-transcribed Cre-recombinase mRNA 27 was transfected into the producer clones. Transfection was performed with Lipofectamine 2000 (2.2 µL/µg mRNA, Invitrogen) according to the manufacturer’s instructions as previously described. 27 Transfected cells were cloned via limiting dilution in 384-well plates. As the clean-up reaction removes the RAGE-unit from the targeted locus, the cells become neomycin sensitive. Thus, selection for positive clones was performed by a split transfer into medium with or without G418 (1,000 ng/mL).
Clean-up was monitored by PCR on genomic DNA with locus-specific primers (Fig. 4A). Genomic DNA was isolated from the producer cells by standard column preparation (QIAamp DNA Blood Mini Kit, Qiagen) and amplified with Phire polymerase (Thermo Fisher Scientific) according to the manufacturer’s instructions, except for the addition of 2.5% (final) formamide (Sigma-Aldrich). The following primers were used: TCU-s (5′-TCCAGGCTCTAGTTTTGAC), CleanL-s (5′-AGCTCGGTACCTTTAAGAC), and the locus-specific antisense oligo FR28-as (5′-GTTTTGTTGGGAGCCTAAGC). PCR cycling protocol was 98°C 4:30 min; 30 cycles of 95°C 30 s, 56°C 60 s, and 72°C 20 s; and 72°C 120 s. Reaction products were loaded on a 1% agarose gel and visualized. For verification of correct targeting and clean-up, the fragments were subcloned into the SK II+ plasmid and sequenced (Eurofins Genomics).
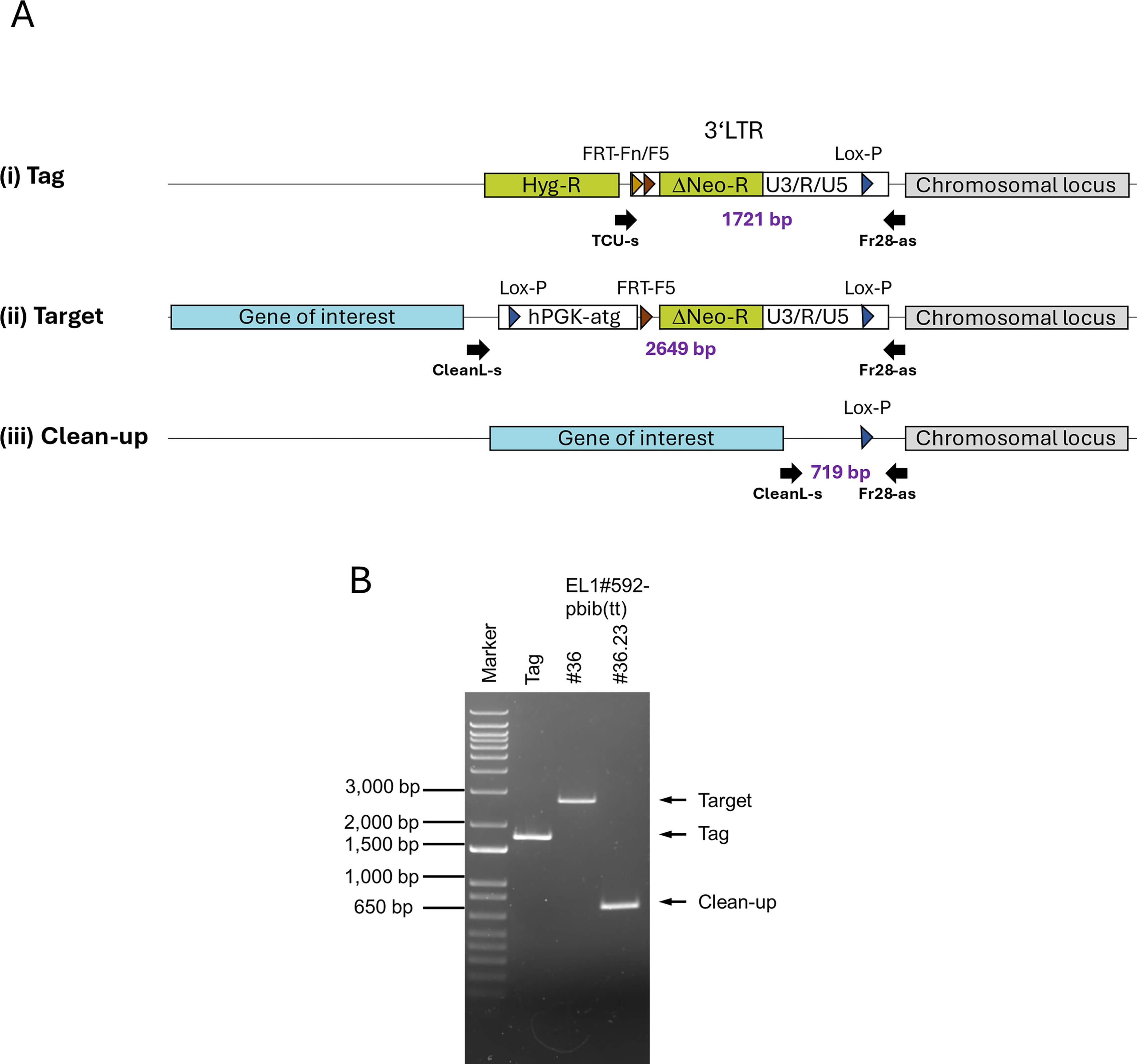
Processing of targeted producer clones.
Up-scaling of vector production
To test the optimal FCS concentration (Supplementary Fig. S3), producer cells were seeded at 4 × 104 cells/cm2 3 days prior to induction with DMEM containing 10% FCS and 100 ng/mL doxycycline. To mimic the volume/area ratio of the HS36 system, 0.2 mL cell culture medium/cm2 was used in a six-well format. Scale-up was implemented from 10 cm to 15 cm tissue culture dishes to a four-layer Nunc Cell Factory System, using identical cell densities for seeding throughout. The growth medium was replaced with harvest medium containing different concentrations of FCS. Titers were determined as described before.
To culture cells in HS36 bioreactors (Corning), cells were seeded in 3.9 L of medium containing 100 ng/mL doxycycline. Cells were induced by removal of the doxycycline-containing medium, washed once with 3.9 L of growth medium, and incubated in harvest medium containing 4% FCS and without doxycycline for 3 days. Supernatant was harvested, filtered (0.45 µm, Cytiva, previously Pall Corporation), aliquoted, and stored at −80°C until further usage.
The Ascent fixed-bed bioreactor system (Corning) was installed according to the manufacturer’s instructions with a 5 m2 fixed-bed bioreactor. Cell seeding was done with 2 × 109 cells (4 × 104 cells/cm2) in a total volume of 3.3 L medium with 100 ng/mL doxycycline and a circulation speed of 250 mL/min with a stepwise decrease of 50 mL/min every 30 min. Cell attachment was monitored by counting the cells left in circulation every 30 min. After successful attachment (>99% of total cells), automated regulation was initiated. The set point for temperature was set at 37°C, pH was set to 7.2, and the level of dissolved oxygen (dO) as the main regulatory parameter for the Ascent bioreactor system was set to 20% at the outlet of the bioreactor.
Cells were induced 3 days after inoculation via complete removal of the expansion medium from the system, followed by one wash with 3 L of fresh medium without doxycycline for 1 h and finally replacement with fresh harvest medium (DMEM, 4% FCS, 3.3 L). After 1 day, perfusion (pH 7.2, 20% dO) was started with a flow-through of 8 L medium per day. Every 24 h, the vector-containing supernatant was harvested, filtered (0.45 µm, Cytiva, previously Pall Corporation), aliquoted, and stored at −80°C until further usage.
TFF was used to purify and concentrate the harvested supernatants. For vector purification, 3.9 L of the harvested supernatant was thawed and concentrated with a TFF module with 750 kDa cutoff (S06-E750-05-S, Repligen) connected to a tubing set (I-SA-P60 95-32-159, Silicone Altimex), and the following steps and parameters: (1) equilibration of the TFF module loop with 3.3 L HEPES-buffer (10 mM HEPES/0.9% NaCl, Sigma-Aldrich), 1.5 L/min flow rate; (2) volume reduction/concentration of the supernatant (170 mL final volume), 2.5 L/min flow rate; (3) diafiltration with 3.3 L HEPES-buffer, 2.5 L/min flow rate; (4) TFF-product 1 (first flush) of 170 mL diafiltered vector-containing medium, 0.25 L/min flow rate; (5) TFF-product 2 (second flush) with 170 mL HEPES-buffer, harvest by gravity flow. During the concentration/diafiltration process, the retentate valves were fully open. The TFF products were pooled, aliquoted, and stored at −80°C until analysis.
Vector-containing medium (before and after TFF purification) was analyzed for p24 protein (Lentivirus-associated p24 ELISA Kit, Cell Biolabs, Inc.) and residual BSA (Serazym Bovine Serum Albumin ELISA Kit, Seramun Diagnostica) according to the manufacturer’s instructions.
Transduction of primary T cells
Primary T cells from healthy volunteers were thawed in serum-free X-Vivo 15 medium (Lonza) supplemented with 5% CTS (Gibco), 1% Glutamax (Gibco), 1% Transact (Miltenyi Biotec), 450 U/mL IL-7 (Cellgenix), and 50 U/mL IL-15 (Cellgenix). Two days after thawing, cells were transduced with an MOI of 2 or 4 of TFF purified LV-containing supernatant in the absence or presence of transduction enhancers. The cell culture dishes were coated with 5 µg/cm2 Retronectin (Takara Bio) for 1 h at room temperature and washed once with phosphate-buffered saline (PBS) prior to transduction. Afterward, cells and viral vectors were added to the coated plates and incubated for 3 days in medium without Transact. Lentiboost (1 mg/mL final concentration, Sirion Biotech) was added to the cells along with the viral vectors for 3 days in medium without Transact. Transduction efficiencies were determined via flow cytometry of eGFP-expressing T cells.
Expression analyses
Western and Northern blot analysis was performed by standard gel and blotting procedures. In brief for Western blotting, cells were harvested and washed three times with 1 mL 1× PBS (centrifugation at 2,500 rpm for 1 min at 4°C) and finally resuspended in 200 µL WB lysis buffer (10 mM Tris-HCl pH 7.4; 100 mM NaCl; 1 mM EDTA; 20 mM Na4P2O7; 1% Triton X-100; 10% Glycerol; 0.1% SDS; 0.5% Na-Deoxycholate, 1× protease inhibitors [Roche]). Cells were lysed for 30 min on ice on an orbital shaker, centrifuged at 14,000 rpm for 10 min at 4°C, the supernatant transferred to a new tube, and stored at −20°C.
Proteins were mixed with loading buffer, separated by 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis, and transferred onto nitrocellulose membranes. Membranes were then incubated overnight at 4°C with anti-p24 rabbit polyclonal antibody (Abcam, ab63913; 1:1,500 in Tris-buffered saline with Tween-20). Detection of gag/pol processing products was performed using CPD-Star (Tropix, Bedford, MA) according to the manufacturer’s instructions after incubation of the membrane with alkaline phosphatase-labeled secondary antibody (Abimed) and analyzed using a ChemiDOC Touch (Bio-Rad).
For non-radioactive northern blotting, in brief, total RNA was extracted from cells by phenol–chloroform extraction, separated on 1% MOPS-buffered denaturing agarose gels and transferred by capillary transfer onto a neutral nylon membrane (Millipore). RNA size markers were from Promega (Mannheim, Germany).
Hybridization was performed at 37°C overnight using a mixture of gene-specific DNA probes directed against HIV-syn.Rev, HIV-syn.GP, and VSV.g. Probes were generated by PCR from the respective plasmids and labeled with biotin during amplification.
Detection was performed using CPD-Star (Tropix, Bedford, MA) as substrate by exposure of the membrane to X-Ray film (Kodak Bio-Max light; Sigma, Munich, Germany). As a loading control, rat GAPDH was used with the same conditions after stripping of the previously developed blot.
Oligonucleotides used for probe synthesis were: rat GAPDH, G-1 (5′-AATGCATCCTGCACCACCAAC), G-2 (5′-GCCATATTCATTGTCATACCAGG); HIV-syn.Rev and VSV.g glycoprotein, both amplified from pBluescript SKII+ subclones; T7 (5′-TAATACGACTCACTATAGG), T3 (5′-AATTAACCCCTCACTAAAGG); HIV-syn.Gag/Pol (size 864 bp), GP864-s (5′-CATAAGGCAAGAGTTTTGG), GP864-as (5′-GAGTATTGTATGGATTTTCAG).
RESULTS
Improved vector design for stable lentiviral packaging cell lines
Current challenges in LV generation include maintaining stable production over time and limited scalability. To address these challenges, we developed reliable lentiviral packaging cell lines, which enable stable target gene integration and, therefore, extended vector production.
First, we designed optimized packaging vectors with reduced risk of RCL formation. We sequence-optimized all viral sequences (VSV.g, rev, tat, gag/pol [GPwt]) to minimize homology of cis-acting elements, improve codon usage and reduce splice sites (see Section “Materials and Methods”) before insertion into the gRV SIN-vector ES.12-6 (Fig. 1A). Additionally, a tet-controlled bidirectional promoter enabled usage of unique 5ʹ and 3ʹ untranslated regions antisense to the gRV genome for gag/pol, VSV.g, and rev. The tet-responsive promoters were modified for high-level expression of LV genes in the antisense context of the vectors.
The optimized syn.GP produced lower vector titers after stable integration than GPwt (Fig. 1B). As expected, the absence of rev resulted in complete loss of titers for GPwt (Fig. 1B) and was accompanied by a strong reduction in gag/pol processing products (Fig. 1C) and decreased levels of reverse transcriptase (RT) and p24 (Fig. 1D). As the syn.GP construct, in contrast to GPwt, did not contain the Rev response element (RRE), we next tested how addition of RRE affected vector titers, RT and p24 levels (Fig. 1B, D). While vector titers increased 2-fold upon addition of RRE to syn.GP, these titers were still 3.5-fold lower than the GPwt titers (Fig. 1B). Parallel measurements of RT and p24 showed a change in the RT/p24 ratio from 1.8 with GPwt to 0.8 with syn.GP, indicating differences in processing (Fig. 1D). As previously reported,16,31 the ratio of RT/p24, rather than their actual levels, indicates clones with high titers. Stepwise reversion of deleted splice sites in non-codon–optimized regions and replacement of a larger area within syn.GP with the wt sequence, creating syn.GP4 (Fig. 1E), restored the RT/p24 ratio to values obtained for GPwt (Fig. 1D). These changes were accompanied by a 2- to 3-fold increase of vector titers (Fig. 1B), gag/pol protein processing comparable to GPwt (Fig. 1C), and were completely independent of the RRE/rev system.
Together, these modifications enhanced the safety of lentiviral packaging cells, especially when combined with single-copy integration.
Generation and characterization of EL1-820 packaging cells
Low copy numbers of expression units improve the reliability of a packaging cell line. We adjusted the multiplicity of infection (MOI) to ensure that the conditional gRV packaging vectors were introduced as single copies, confirmed by target locus amplification (TLA)-based sequencing (Supplementary Table S1).
Figure 2A illustrates the stepwise generation of the final producer line. A previously selected neomycin sensitive 293T clone was transduced with ES.12-6(TeG).ps to introduce stable, constitutive expression of a 4C-specific tet-responsive transactivator, tTA4CE (tet-OFF32,33). Single cell clones were selected via MAXTA and limiting dilution and tested for LV production by transient transfection of five plasmids (VSV.g, rev, tat, gag/pol, and the LV-transfer plasmid pRRL-SIN.ppt.PGK.GFP.WPRE 29 ) to finally yield clone EL1-t.
Next, we generated the final stable packaging cell line by stepwise introduction of the MT vectors (Fig. 1A) into the EL1-t clone (Fig. 2A), resulting in the clonal cell lines EL1-tr#17 (containing tTA4CE and syn.Rev; puromycin selection), EL1-trg#23 (tTA4CE, syn.Rev, and syn.GP4; zeocin selection), and EL1-trgv#81 (tTA4CE, syn.Rev, syn.GP4, and VSV.g; Diphtheria toxin A selection). At each step, single-cell clones were screened for production capacity by titrating supernatants generated by transient expression of the missing components from plasmids (Fig. 2B, C) and advantageous phenotypes (i.e., low syncytia formation, fast growth). Northern blot analysis of the generated clones showed that expression of all three stably integrated genes was strictly dependent on tet-controlled induction (Fig. 2D). Expression of VSV.g resulted in about 80% reduction of cell growth compared with uninduced cells (Fig. 2E), while expression of syn.Rev or syn.GP4 did not alter proliferation. The formation of syncytia after induction of the integrated packaging genes was minimal (not shown). Together, these results suggest that the EL1-trgv#81 packaging clone is suitable for prolonged vector production.
Titers of EL1-trgv cells were determined under stable conditions by a specifically designed, LTR-driven lentiviral test vector (LCO, Fig. 2F). This vector was necessary for stable testing to determine the production potential of the cells and if the vector genome was integrated only as a single copy. To investigate whether HIV-tat is important for vector production after single-copy integration, LCO was designed either with, LCO(tateGB)ps, or without, LCO(GB)ps, co-expression of HIV-tat. After blasticidin selection, EL1-trgv cell clones (#60, 81, and 95) were tested for vector production. In the presence of HIV-tat, at least 10-fold higher titers (2–3 × 106 TU/mL) were reached by EL1-trgv#81 (Fig. 2G).
In summary, we have generated cell lines with stable, inducible integration of the components for LV production with a vector yield comparable to transient transfections.
FRT-integration and selection of a tagged packaging cell line
Stable LV production requires integration of its expression cassette into a packaging cell; however, vector genome expression is largely dependent on its integration site, and hence producer clone selection requires cumbersome screening efforts. To overcome this limitation, we took advantage of RMCE technology 34 to generate customized producer clones. The genomic tag (FRT-Fn and FRT-F5 sites) necessary for site-directed introduction of the vector expression cassette was introduced into EL1-trgv#81 via the previously described gRV tagging vector ETAG.fcN 27 (Fig. 3A). Clones (EL-820) were selected by limiting dilution and hygromycin selection (Supplementary Fig. S1). A screen was performed to identify clones with a suitable integration site for LV-vector production based on site-directed recombination-dependent activatable gene expression (RAGE) of the neomycin resistance gene (Supplementary Fig. S2). About 700 EL1-820 clones were tested by targeting with a packageable LV-vector and subcloning a small progeny of three to five clones each for characterization. Testing constructs pbib-, pbib(t)-, and pbib(tt)-ELTAR.fc-pRRL (Fig. 3B) were designed to transfer a single-copy SIN-vector expression cassette. Because of encouraging results from the LCO-based screen (Fig. 2G), testing constructs expressed the vector genome either constitutively or under tet-control, and additionally with or without syn.Tat expressed in the opposite direction.
Following RMCE of pbib-ELTAR.fc-pRRL (Fig. 3B), up to 3 × 106 TU/mL of LV-SIN-vectors were generated from a single productive vector copy integrated at the tagged locus. Only few clones complied with our requirements, including a shift from red to green fluorescence after replacement of mCherry with GFP, high titer, and favorable growth characteristics (e.g., reduced syncytia formation). Based on this initial screen, we chose four tagged packaging clones (EL1-820#1, #10, #333, and #592) for further analysis.
Titers from a single-copy integrate of an LV SIN-vector via RMCE were approximately 3 × 106 TU/mL, thus at least 10-fold below titers routinely achieved by transient production for this vector. Because HIV-syn.Tat co-expression led to a substantial vector titer increase (as seen with LCO(tateGB)ps; Fig. 2G), we introduced HIV-syn.Tat into the antisense-operating constitutive expression unit of pbib-ELTAR.fc-pRRL via T2A-coupling to the GFP/blasticidin selection marker. After targeting the resulting exchange construct (pbib(t)-ELTAR.fc-pRRL; Fig. 3B) into EL1-820#592, titers in three of four tested clones increased (Fig. 3C). For the finally selected EL1-820#592 packaging cell clone, titers of about 2.5 × 106 TU/mL without HIV-syn.Tat expression were determined, while titers increased about 3-fold to 7.1 × 106 TU/mL with syn.Tat (Fig. 3D). However, clones with syn.Tat expression showed reduced growth capacity (Fig. 3E), presumably due to squelching.35,36 We therefore replaced constitutive tat expression with an inducible bidirectional promoter, resulting in pbib(tt)-ELTAR-pRRL (Fig. 3B). In construct pbib(tt)-ELTAR-pRRL, the strong Ptet.T3-promoter controls vector genome expression, while the weak Ptet.T8-promoter drives HIV-syn.Tat expression.28,37 Introduction of this exchange construct into the same locus of EL1-820#592 packaging cells restored proliferation (Fig. 3E), and increased vector production about 3- to 5-fold (1–2 × 107 TU/mL; Fig. 3D) for up to six consecutive harvests starting 72 h after induction (Fig. 3F). Thus, efficient therapeutic vector production from a single-copy integrate was restored to the level of transient transfection by tet-controlled co-expression of the vector genome and HIV-syn.Tat.
Processing of targeted producer clones
Introducing the targeting construct and selecting for correct LV-vector expression via RMCE leaves residual ETAG.fcN sequences containing the 3ʹ-LTR and RAGE-unit (Fig. 4A), which can produce therapeutic LV-SIN-vectors capable of conferring neomycin resistance to target cells. The ratio of recombined to correct vectors is roughly 1 to 1–5 × 104 in our system, necessitating the removal of contaminating LVs for clinical vector production. Transfection with Cre-recombinase messenger RNA (mRNA) excised the RAGE-unit along with the residual LTR. Genomic PCR developed to amplify locus-specific fragments confirmed sequence removal (Fig. 4B).
Up-scaling of vector production
The clinical application of gene therapeutic treatments requires large numbers of LVs and thus high quantities of producer cells. The scalable Hyperstack system provides the identical surface as the cell culture dishes used to establish the process and increases the production area up to 1.8 m2 in one Hyperstack 36 bioreactor (HS36).
The HS36 bioreactor was inoculated with about 7.5 × 108 producer cells (4 × 104 cells/cm2) expanded from one vial of EL1-820#592-pbib-(tt)#36.21 (clean-up clone) in the off-state (+dox, 100 ng/mL). Three days post-seeding, cells were induced by dox removal. Fetal calf serum (FCS) concentration during production was reduced to 4% to lower costs and minimize clogging of the filter during downstream purification, with no significant decrease in titers during several days of induction (Supplementary Fig. S3). Four days post-seeding (24 h post-induction), harvesting was started at 24 h intervals. Across four independent production runs, vector titers increased within 72 h post-induction to a maximum value of around 1 × 107 TU/mL (days 6–11) and then slowly decreased (Fig. 5A, B). The total amount of transforming units harvested at each timepoint (3.9 L) remained above 1 × 1010 TU for days 6–13, resulting in a mean yield for one production run of about 2.3 × 1011 TU.
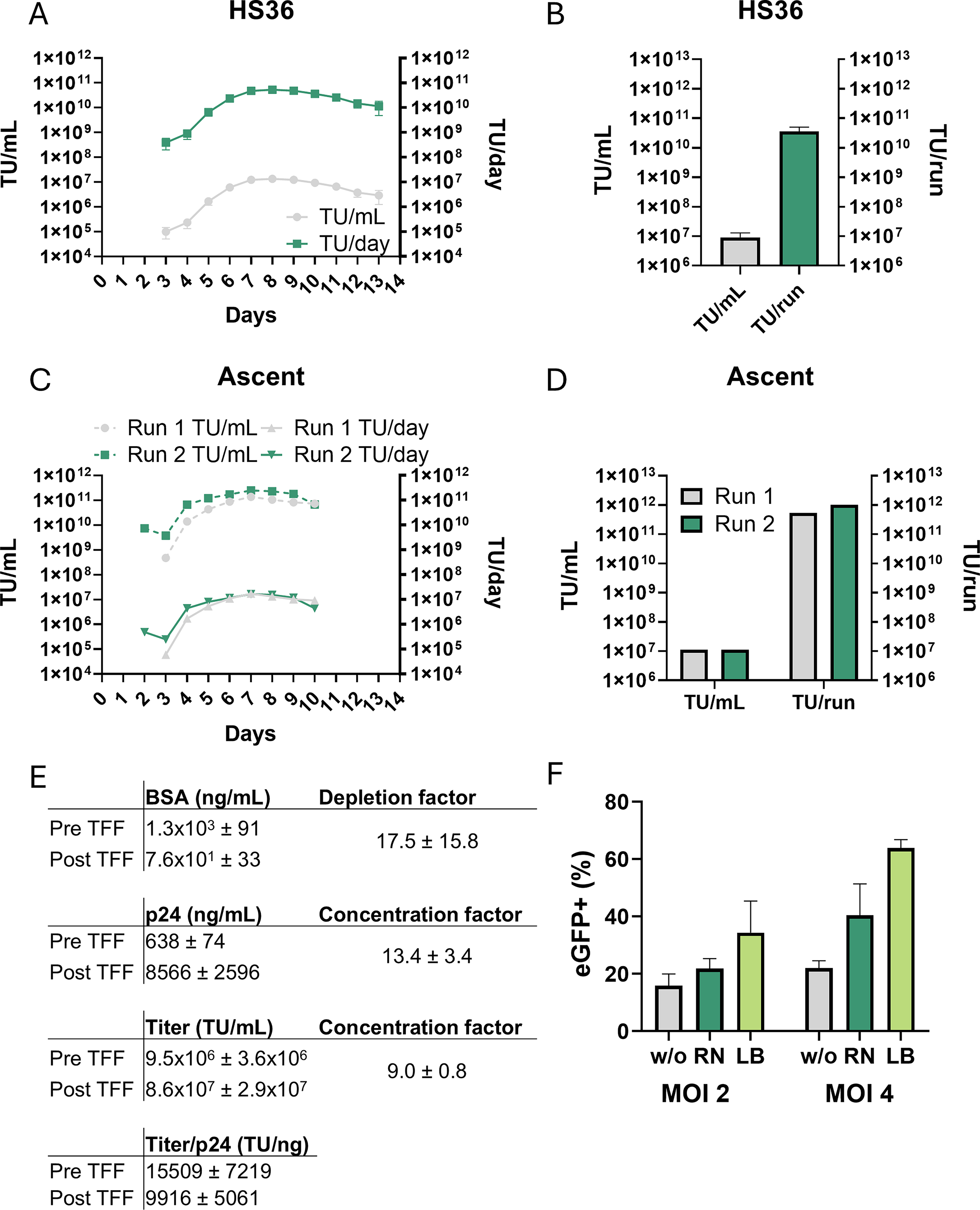
Up-scaling of vector production.
To further increase the system’s production capacity, we evaluated the novel fixed-bed Ascent bioreactor system, a closed system that contains a similar matrix for cell attachment as the HS36 bioreactor but allows strict control of growth parameters. Two independent test runs (Ascent 1/2) were performed with a 5 m2 bioreactor, that is, approximately 2.8-fold larger compared with HS36. Cell seeding and induction were performed analogously to the HS36 system. Medium exchange/harvest was done in batches with 8 liters of new medium every 24 h for at least 6 days. For the second run, the harvest volume was increased to 15 liters per 24 h since the glucose concentration in the first run decreased to 6 mg/dL at Day 6. Kinetics of vector production in the Ascent system for both runs were similar and comparable to the HS36 system (Fig. 5C, D). Interestingly, titers produced by an almost doubled volume of medium in the Ascent run 2 indicated that cells require a certain level of glucose (e.g., > 50–100 mg/dL) for constant high production. This increase in medium resulted in an overall increase of vector yield to up to 1 × 1012 TU (1.4 × 107 TU/mL and 283.6 TU/cm2/mL) for the second production run.
Next, we established tangential flow filtration (TFF)-based downstream processing of the crude harvested supernatants. Each harvest was filtered (0.45 µm) to remove cell debris and stored at −80°C until further processing. To demonstrate the feasibility of production/purification, the harvests of Day 6 (3.9 L) from five different production runs from the HS36 system were thawed, independently purified, and concentrated via TFF. The results (Fig. 5E, Supplementary Fig. S4) showed that the vectors were concentrated about 9-fold with a recovery of around 80%. Determination of p24 concentration revealed about 10,000 TU/ng p24, while serum-based bovine serum albumin (BSA) was reduced around 17-fold (to 77 ng/mL).
One production run of the processed vectors was finally tested on primary T cells with or without transduction enhancers (Retronectin or Lentiboost). Transduction efficiencies of 16% (MOI 2) and 23% (MOI 4) were achieved without transduction enhancers (Fig. 5F). Retronectin increased transduction rates to 20% (MOI 2) and 39% (MOI 4), respectively, while Lentiboost showed the highest efficiencies with 40% (MOI 2) and up to 65% (MOI 4) (Fig. 5F).
Collectively, our stable producer clones can maintain large-scale production of functional vectors capable of transducing primary T cells after TFF purification.
Production of a putative therapeutic vector
Finally, we evaluated the suitability of our system for the production of clinically relevant, stable therapeutic LV SIN-vectors by generating a lentiviral targeting construct expressing a chimeric antigen receptor (CAR) targeting the tumor-associated antigen Claudin-6 (Fig. 6A). 38
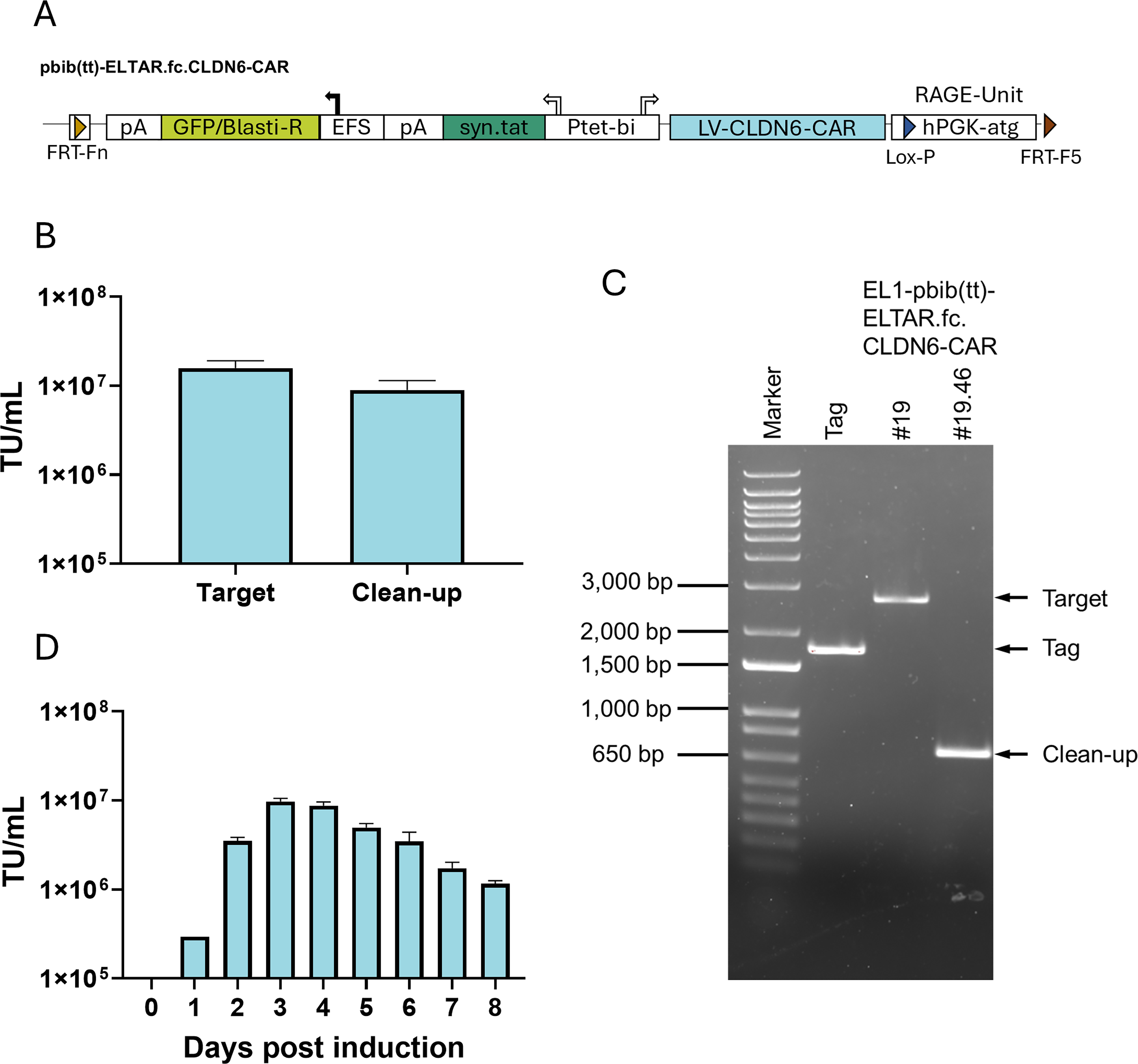
Production of a putative therapeutic vector.
We stably integrated the CLDN6-CAR plasmid pbib(tt)-ELTAR.fc.CLDN6-CAR via RMCE into EL1-820#592 cells to generate the producer clone EL1-pbib(tt)-ELTAR.fc.CLDN6-CAR. Upon dox removal, titers of around 1 × 107 TU/mL were achieved in six-well plates (Fig. 6B). We excised the RAGE-unit by Cre-recombinase mRNA transfection and demonstrated successful clean-up by genomic PCR (Fig. 6C). We observed similar induction kinetics as in the previously tested GFP-transferring LV SIN-vector, peaking 72 h post-induction and slowly decreasing thereafter (Fig. 6D).
In summary, we present a reliable, stable cell line for robust production of custom LVs at large scale. Our system is suitable for efficient production of clinical-grade vectors and therefore provides a more cost-effective solution with less variation between batches.
DISCUSSION
Here, we describe several improvements for the stable production of LVs. We developed EL1-820 cells containing all essential packaging components stably integrated and enabling the introduction of the vector expression cassette by RMCE into a single, specified site. Viral titers were comparable to transient transfection, and scaled-up production was successfully tested in two bioreactor systems.
This work builds on previous efforts to create stable producer lines,6,7,10 incorporating advances in codon usage optimization, translational efficiency, 26 structural requirements, 25 and the balanced ratios of packaging components. 10 Most lentiviral transfer vectors used in clinical trials today contain the native HIV-1 packaging region that reaches into wt-gag. In addition, a particular region of wt-pol contains the CTS and the central polypurin tract. Both regions are indispensable for the generation of transfer vectors but pose a high risk for homology-dependent recombination with wild-type gag/pol in production systems. Therefore, the reduction of sequence homology in these regions of gag and pol is mandatory for efficient and safe generation of LVs and was incorporated into our system. Finally, improved tet-regulated promoters37,39 allowed us to generate a high-titer packaging cell line with single-copy expression of each packaging component transferred via gRV SIN-vectors.
One major upgrade here is the application of RMCE technology.27,30,40 In existing producer cell lines (e.g., GPRG 7 ), a concatemeric design to integrate the expression unit was used. Introduction of large concatemeric repeat regions can lead to rearrangements, which may destabilize the integration and thus affect vector production. In our system, a single copy of the expression unit is integrated directionally into a predefined genomic locus, making vector production predictable, highly stable, and cost-efficient due to decreased batch testing and less material needed per production run. Moreover, single-copy integration enables integrity verification by TLA-sequencing, important for regulatory compliance.
RMCE further allows clone selection independent of transgene or vector expression via RAGE, enabling marker-free vector production. After targeting, the RAGE-unit and residual gRV sequences are removed, eliminating contaminating LVs from recombination with tagging vector sequences (i.e., neoR-transfer), providing a stable, contamination-free producer cell line.
Notably, the HIV-tat gene was important to achieve higher titers (Fig. 2G), likely due to its transcription-enhancing capabilities. However, the constitutive overexpression of transactivators, for example, syn.Tat, can sequester transcriptional cofactors (squelching) and thereby decrease the expression of important genes, eventually leading to growth reduction.35,36 This growth reduction could be mitigated with an inducible expression system (Fig. 3E).
Selected clones remained stable over a period of four months without drug selection, except for doxycycline to keep expression of tet-controlled units in the “off-state.” Large-scale production in HS36 and Ascent bioreactor systems yielded large numbers of vectors (2.3 × 1011 TU and 1 × 1012 TU per run, respectively). Together with subsequent TFF purification, we achieved up to 60% transduction efficiency in primary T cells, providing evidence that the generated vectors are functional. Finally, we successfully generated a putative therapeutic SIN-vector (CLDN6-CAR) with titers comparable to those of our test vectors. This demonstrates that our system can produce gene therapy vectors at a relevant scale while reducing costs by eliminating the need for transient transfections.
In summary, EL1-820 is a robust, customizable cell line for clinical-grade LV production. Its site-specific, single-copy integration supports safe, scalable, and predictable generation of VSV.g-pseudotyped lentiviral SIN-vectors for gene therapy.
AUTHORS’ CONTRIBUTIONS
K.K. and R.L. conceptualized the work. N.H. and R.L. planned, supervised, and analyzed the experiments. N.H., R.L., K.Z., L.C.C., L.M., and S.O. cloned the used constructs. K.Z., L.C.C., L.M., S.O., F.R., M.N., and M.H. tested the generated cell lines. F.R., M.N., and M.H. established production scale-up and TFF purification. N.H. and R.L. wrote the article.
Footnotes
ACKNOWLEDGMENT
The authors would like to thank Luigi Naldini for providing the expression plasmids for the transient production of LVs. They would like to thank Corning for providing them with the Ascent bioreactor prior to its market release and the continuous support during the setup of vector production. Furthermore, they would like to thank Florian Bock (BioNTech SE) for providing medical writing assistance.
FUNDING INFORMATION
The work presented here was funded by BioNTech-IMFS.
DATA AVAILABILITY
Data are contained within the article or supplementary material. Detailed sequences of the used constructs will be provided to customers and authorities upon request.
AUTHOR DISCLOSURE
N.H., L.C.C., K.Z., F.R., M.N., M.H., S.O., and R.L. are employees of BioNTech-IMFS (Germany). L.M. and K.K. are former employees of BioNTech-IMFS (Germany). R.L. owns stock and/or stock options in BioNTech SE. R.L. serves as a consultant for TET Systems.
Supplemental Material
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.