
Abstract
Gene therapy is emerging as a transformative approach for treating amyotrophic lateral sclerosis (ALS), a progressive and fatal neurodegenerative disease. While gene replacement has shown a groundbreaking success in spinal muscular atrophy, the complexity of ALS—due to frequent gain-of-function mutations and a heterogeneous etiology—presents significant challenges. Importantly, approximately 90% of ALS cases are sporadic, with unknown genetic mutation, further complicating patient stratification and therapeutic targeting. As a result, gene therapy strategies must often address multiple pathological mechanisms simultaneously. So far, current gene therapy strategies aim to either suppress toxic gene expression or promote neuroprotection, predominantly via viral-mediated delivery systems. This review will provide an overview of emerging preclinical and clinical gene therapy approaches for ALS, focusing on two main strategies: gene silencing and neuroprotection. Gene silencing techniques, including antisense oligonucleotides (ASOs), viral-mediated RNA interference, and gene editing, have demonstrated efficacy in reducing mutant gene expression, particularly in SOD1 and C9orf72 models, although clinical translation has so far yielded limited success. The recent Food and Drug Administration’s approval of the ASO therapy Qalsody for SOD1-ALS underscores the clinical potential of these approaches. Neuroprotective strategies aim to enhance motor neuron survival through delivery of trophic factors, often targeting both central and peripheral tissues to harness retrograde transport mechanisms. We will discuss the advantages and limitations of various delivery vectors, targeting specificity, timing of intervention, and translational challenges, alongside current clinical trial data. This review aims to synthesize how these approaches may converge to address the multifaceted nature of ALS and guide the development of next-generation therapeutics.
INTRODUCTION
Therapeutic landscape of amyotrophic lateral sclerosis
Gene therapies offer significant potential in addressing rare, devastating, and life-threatening diseases such as amyotrophic lateral sclerosis (ALS), as they usually focus on correcting the underlying genetic cause of the condition rather than just alleviating the symptoms. While gene therapy—including vector-based strategies, antisense oligonucleotides (ASOs), and RNA interference—has recently gained significant attention, a range of nongenetic strategies have also been explored preclinically in ALS. These include small molecules, peptide- or protein-based systems, and stem cell-derived therapies. Currently, only three nongenetic treatments are approved for ALS: riluzole, edaravone, and Relyvrio. Riluzole, the first approved drug, likely acts via antiglutamatergic mechanisms but offers only a modest survival benefit of 3–4 months. 1,2 Edaravone, which is thought to be a free radical scavenger, showed efficacy only in a narrow patient subgroup and was later withdrawn in Europe due to insufficient benefit. 3 Finally, clinical trials of Relyvrio suggest a modest delay in disease progression by reducing endoplasmic reticulum (ER) and mitochondrial stress. 4,5
The notable success in treating infants with spinal muscular atrophy (SMA) underscores the promise of gene therapy for other neuromuscular disorders and motoneuron diseases (MNDs). However, unlike SMA, ALS is uncommonly a monogenic disorder, complicating the development of therapeutic strategies. Additionally, in cases with a genetic basis, the disease is most often associated with gain-of-function mutations, rendering gene replacement therapies unfeasible. Nevertheless, numerous gene therapy strategies have been investigated in preclinical settings, primarily focusing on gene supplementation with factors capable of mitigating ALS pathological alterations (outlined in Table 1). These approaches commonly involve the direct delivery of viral vectors—such as adeno-associated (AAV), adenoviral (Ad), or lentiviral (LV) systems—or the transplantation of genetically engineered cells. In parallel, the targeted suppression of pathogenic genes has been extensively explored using ASOs and viral-mediated knockdown via short hairpin RNAs (shRNAs) or microRNAs (miRNAs) (summarized in Table 2). Notably, clinical trials employing these approaches have shown promise, particularly targeting SOD1 mutations (Qalsody), with ongoing evaluations directed to FUS (NCT04768972) and C9ORF72 (NCT04931862 and NCT03626012) mutations.
Gene Delivery Strategies Tested in ALS Models
Treatment ages: asymptomatic (Asym), neonatal (N), presymptomatic (Pre), symptomatic (Sym), early symptomatic (ES), and late symptomatic (LS).
AAV, adeno-associated virus; AMPA, α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid; BB, biceps brachii; BBB score, Basso–Beattie–Bresnahan locomotor rating score; CB, cytomegalovirus enhancer/β-actin promoter; Cb, combined; CBA, chicken beta-actin; CBE, cytosine base editor; CMAP, compound muscle action potential; CMV, cytomegalovirus promoter; CNS, central nervous system; CRISPR, clustered regularly interspaced short palindromic repeats; CT-1, cardiotrophin 1; CTNF, ciliary neurotrophic factor; DAO, d-amino acid oxidase; DCN, deep cerebellar nuclei; DIA, diaphragm; FC, facial muscles; FL, forelimb; FMN, facial motor nucleus; G-CSF, granulocyte-colony stimulating factor; GDNF, glial cell-derived neurotrophic factor; GLT1, glial glutamate transporter 1; GLU, gluteus maximus; GluA2, subunit of the AMPA receptor; GM, gastrocnemius; HGF, hepatic growth factor/hepatocyte growth factor; HL, hindlimb; HP, hippocampus; i.c., intracerebral; ICap, internal capsule; i.c.b., intracerebellar; i.c.m., intracisterna magna; i.c.v., intracerebroventricular; IGF-1, insulin-like growth factor; i.l., intralingual; InC, intercostal muscles; i.p., intrapleural; iPSCs, induced pluripotent stem cells; i.s., intraspinal; i.s.t., intrastriatal; LV, lentivirus; MC, motor cortex; MIF, macrophage migration inhibitory factor; MMP, measurement of mitochondrial function; MNs, motoneurons; MPC, muscle progenitor cells; MSC, mesenchymal stem cells; MuSK, muscle-specific receptor tyrosine kinase; N/A, not applicable; NAb, neutralizing antibodies; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; NHP, nonhuman primate; NMJ, neuromuscular junction; NPC, neural progenitor cells; N/R, not reported, NRG1-I, neuregulin type I; NRG1-III, neuregulin 1 type III; NS, neurological score; NT-3, neurotrophin-3; PD, pharmacodynamics; PGK1, phosphoglycerate kinase-1; PhMNs, phrenic motoneurons; PK, pharmacokinetics; pmn, progressive motor neuronopathy; p-NF, phospho-neurofilament; PSM, paraspinal muscles; QD, quadriceps; RSV LTR, Rous sarcoma virus long terminal repeat promoter; SD, Sprague–Dawley rats; SFP, spontaneous fibrillation potentials; SYN, synapsin I; SYT13, synaptotagmin 13; T Age, Treatment Age; TA, tibialis anterior; TB, triceps brachii muscles; TC, thoracic muscles; TG, tongue; t.p., transplant; TS, triceps surae muscle; TU, transducing units; vg, viral genomes.
Gene Silencing and Editing Strategies Tested in ALS Models
Treatment ages: asymptomatic (Asym), neonatal (N), presymptomatic (Pre), symptomatic (Sym), early symptomatic (ES), late symptomatic (LS).
ASO, antisense oligonucleotide; shRNA, short hairpin RNA; siRNA, small interfering RNA.
Gene therapy approaches have focused on targeting either motoneurons (MNs) or the skeletal muscles and their synaptic connections. Local MN pools can be directly reached through intraspinal (i.s.) injections for lower MNs and intracerebral (i.c.) injections for upper MNs. Alternatively, MNs can be broadly accessed through cerebrospinal fluid (CSF) infusion via intrathecal (i.t.), intracisternal magna (i.c.m.), or intracerebroventricular (i.c.v.) routes. To deliver therapeutics to the muscles, strategies have involved intramuscular (i.m.) or intravenous (i.v.) administrations, complemented with the use of retrograde viral vectors or transgenic proteins that, upon expression, will be taken up at the synapse (Fig. 1).
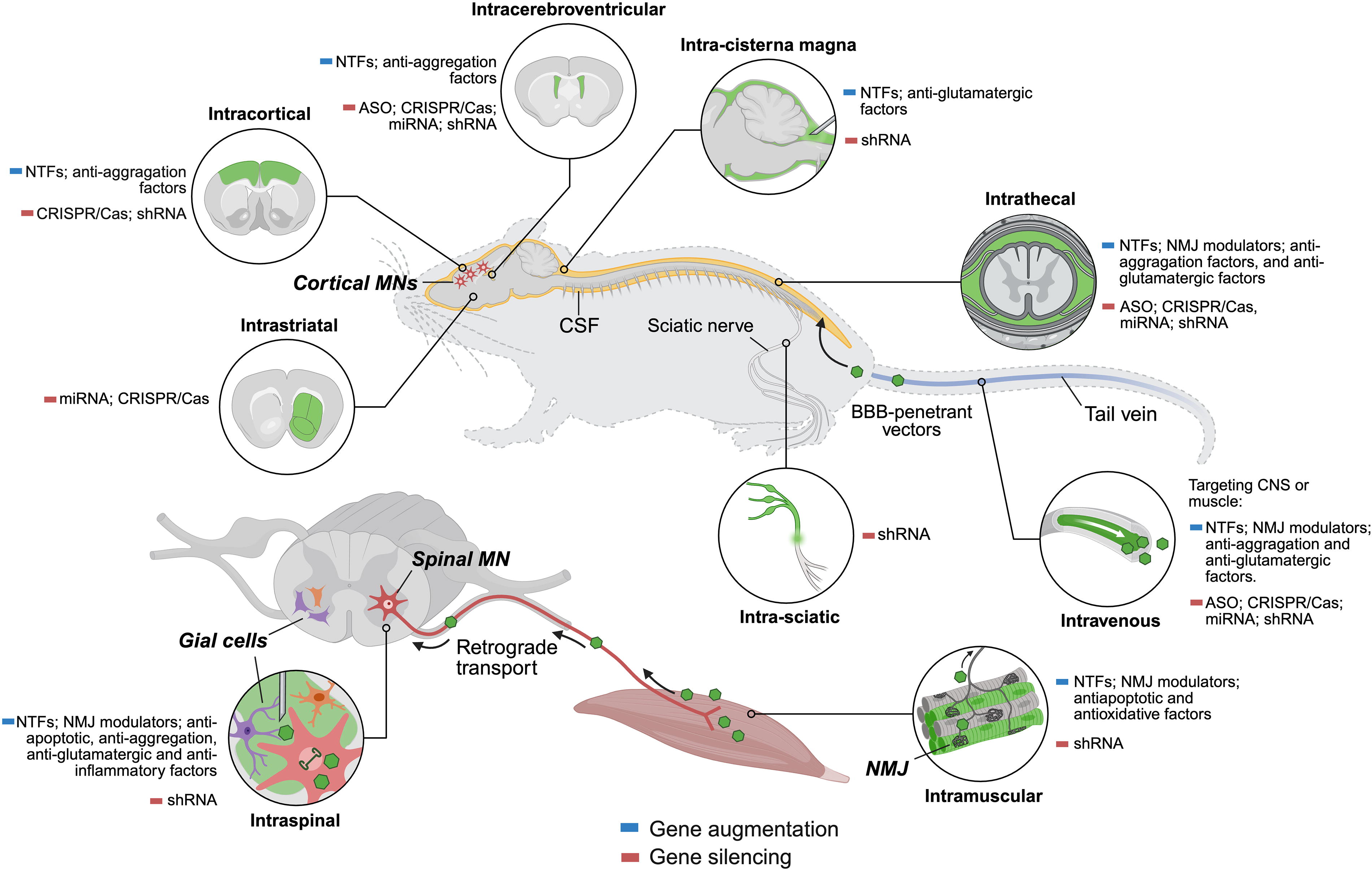
Gene therapy strategies tested in ALS: delivery routes and target cells and tissues. A range of gene supplementation and silencing approaches have been explored in ALS preclinical models, leveraging intraspinal, intracerebral, intrathecal, intracisternal magna, intracerebroventricular, or intramuscular delivery routes. Created in BioRender (https://BioRender.com/ynj06t6). Verdés, S. (2025). ALS, amyotrophic lateral sclerosis; ASO, antisense oligonucleotide; BBB, blood–brain barrier; CNS, central nervous system; CRISPR/Cas, clustered regularly interspaced short palindromic repeats/CRISPR-associated protein; CSF, cerebrospinal fluid; miRNA, microRNA; MN, motor neuron; NMJ, neuromuscular junction; NTFs, neurotrophic factors; shRNA, short hairpin RNA.
In this review, we explore the current landscape of gene therapy strategies for ALS, examining preclinical and clinical approaches, delivery methods, and the challenges that arise from the disease complexity.
Pathophysiological mechanisms and experimental animal models
The identification of ALS-associated genes has enabled the generation of transgenic models replicating key features of the disease, uncovering the pathogenic events underlying MN degeneration and serving as essential tools for preclinical assays. An ideal rodent model for ALS should recapitulate ALS phenotypes and pathologies, particularly involving motor deficits and MN degeneration. As the first gene discovered to be causative of ALS, transgenic mice overexpressing mutant forms of the human SOD1 gene remain the most widely used and best-characterized models. 118 The first ALS animal model featured a high copy number of the human mutant SOD1 gene (mSOD1) with a glycine-to-alanine transition at the 93rd codon. 119 These mice start motor signs around 12 weeks of age, developing trembling, hindlimb weakness, and muscle atrophy, culminating in paralysis and death by 16–20 weeks, which is preceded by axonal degeneration and neuromuscular junction (NMJ) denervation. Histopathological characteristics of the most common profile of patients with ALS, with MN loss accompanied by glial reactivity, except for the absence of mislocalization and accumulation of TDP-43 inclusions are also replicated in these mice. 120 Alterations in SOD1 protein have also been found in sporadic patients with ALS, thus increasing the interest of this murine model. 121 Other SOD1 mutants (e.g., G37R and G85R) and low-copy-number G93A lines have been developed, generally displaying slower disease progression.
Beyond SOD1, additional genetic models have provided insight into ALS pathophysiology. The main pathological finding in most sporadic and familial ALS cases is the presence of ubiquitinated inclusions of TDP-43 in the cytoplasm of neurons and glia in the brain and spinal cord. Models for TARDBP mutations have been generated with wild-type or mutant TARDBP sequences showing ranging degrees of MN degeneration and phenotypes, although most of these models fail in reproducing the ALS motor involvement. 122 The initial Prp-TDP-43 mouse models show cortical and descending corticospinal tract pathology before developing lower MN degeneration. A recent knock-in TDP-43Q331K model recapitulates both ALS and frontotemporal dementia (FTD)-like phenotypes, including reduced muscle mass and impaired motor function, being the most physiological TDP-43 model to date. 123
The C9ORF72 G4C2 hexanucleotide repeat expansion is the most common genetic alteration in patients with ALS and is believed to induce toxicity through gain-of-function mechanisms including RNA foci, sequestration of RNA-binding proteins, and repeat-associated non-ATG translation leading to toxic aggregates of dipeptide repeat proteins (DRPs). Mouse models expressing this repeat recapitulate distinct disease-related pathological, functional, and behavioral phenotypes, 124 although many fail to exhibit TDP-43 pathology, whereas others fail to induce MN loss and severe motor alterations. 125 –127
Mutations in the gene encoding profilin 1 (PFN1), a cytosolic protein that participates in the assembly of filamentous actin, have been identified in several familial ALS cases. PFN1 mutations were modeled in mice (e.g., G118V or C71G) reproducing many key ALS features, including lower and upper MNs loss, abnormal protein ubiquitination, reduced choline acetyltransferase expression, gliosis, muscle atrophy, impaired motor performance, and reduced survival. 90,128,129
Despite the diversity of genetic models, mSOD1 mice containing the G93A mutation are the most used in ALS preclinical research due to their well-defined and reproducible ALS phenotype, which for other models generally only becomes apparent at an advanced age. 118,130 However, the poor translation of therapeutic efficacy from mSOD1 mice to positive results in clinical trials has raised concerns about its representativeness. With more accumulated experience, future preclinical assays should incorporate more than one model reflecting other ALS-linked mutations to better predict clinical outcomes. Given that SOD1 mutations account for <2% of ALS cases, most therapeutic approaches—beyond those blocking the defective SOD1 gene—aim to be generalized to sporadic cases by demonstrating delay in motor deficits and MN degeneration that are the essential alterations in ALS and related diseases.
Mechanistically, these models have been useful to describe a wide variety of cellular pathways contributing to MN degeneration, including glutamate excitotoxicity, oxidative stress, mitochondrial dysfunction, protein misfolding and impaired protein homeostasis, RNA metabolism defects, ER stress, deficits of axonal transport, and neuroinflammation (Fig. 2). These mechanisms may not be mutually exclusive, although it remains unclear whether a unique event triggers all these processes. This topic has been the focus of several review articles and here we only include a brief summary. 131 –136
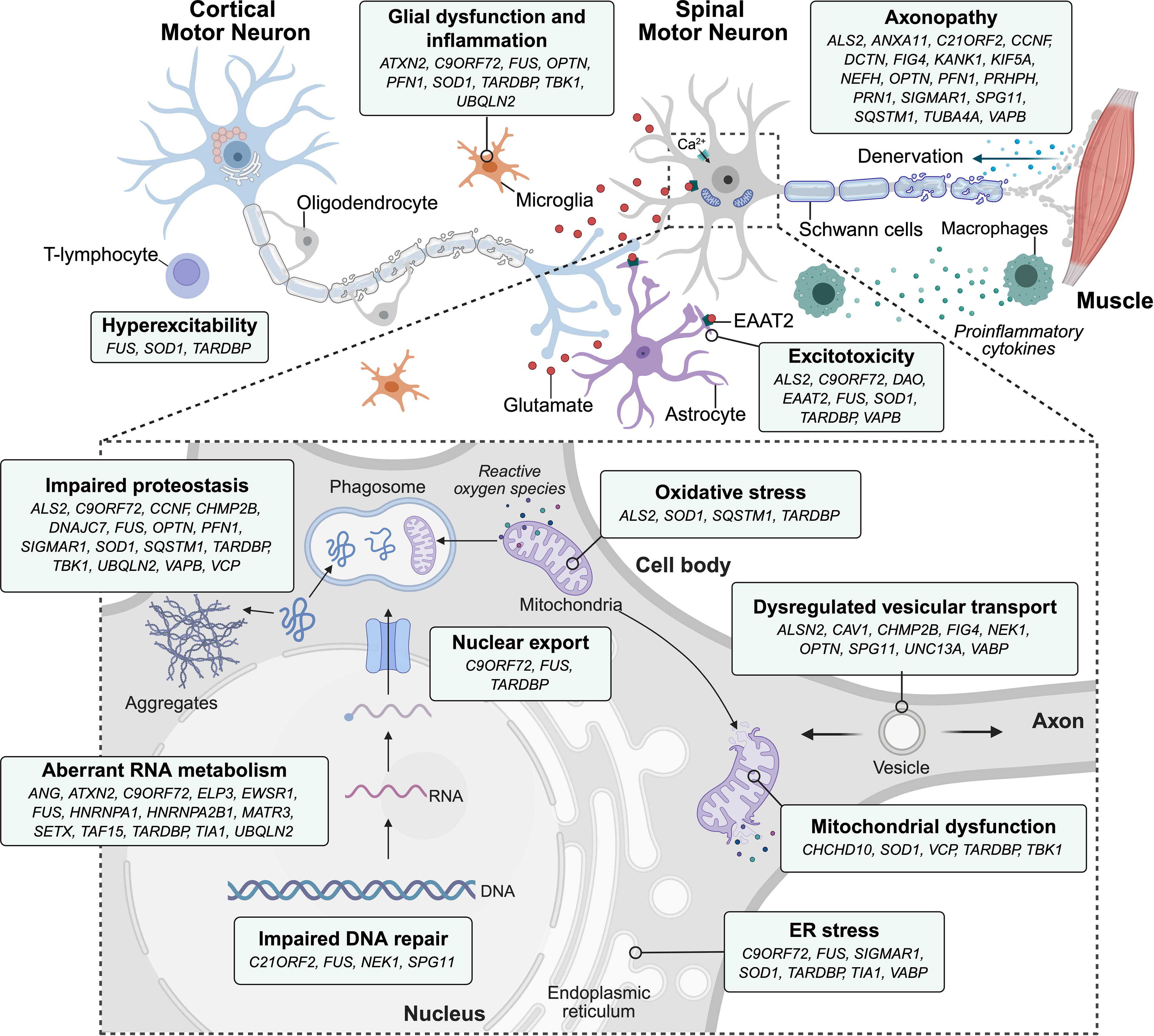
ALS mechanisms and associated genes. Multiple molecular and cellular pathways contribute to motor neuron degeneration in ALS, with many ALS-associated genes (e.g., SOD1, C9ORF72, TARDBP, and FUS) involved in overlapping mechanisms. Key pathological processes include excitotoxicity, glial dysfunction, inflammation, oxidative stress, impaired proteostasis, mitochondrial dysfunction, aberrant RNA metabolism, endoplasmic reticulum (ER) stress, DNA repair deficits, and axonopathy. Main genes linked to each mechanism are indicated in the corresponding boxes. Created in BioRender (https://BioRender.com/fusu0u5). Verdés, S. (2025).
Enhanced excitatory transmitter’s input induces a massive calcium influx into the cytoplasm that damages the cells through the activation of calcium-dependent proteases, lipases, and nucleases. MNs are particularly sensitive due to low calcium buffering capacity and highly permeable α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid (AMPA) receptors. Elevated glutamate levels in CSF of patients with ALS, overactivity of NMDA and AMPA receptors, and reduced astrocytic glutamate uptake support this mechanism. The first drug approved for the treatment of ALS, Riluzole, mainly acts by reducing excitotoxicity. Excitotoxicity has also been linked to mutations in genes such as GRIA2, DAO (d-amino acid oxidase), and SLC1A2 (excitatory amino acid transporter 2, EAAT2), which affect glutamate receptor function and astrocytic glutamate clearance.
Oxidative stress damage to proteins, lipids, and DNA also occurs in patients with ALS, as well as in ALS mouse models. Reactive oxygen species are increased in the CSF and serum of patients with ALS. Mitochondrial abnormalities—such as respiratory chain complex deficits—have been described in the spinal cord and in skeletal muscle of patients with ALS. Experiments performed in mSOD1 mice also revealed early mitochondrial abnormalities prior to symptoms onset. DPRs in C9ORF72-associated models also compromise mitochondrial function and cause oxidative stress and DNA damage. Other genes such as CHCHD10, OPTN, and VCP have been implicated in mitochondrial dysfunction and redox imbalance, further supporting mitochondrial stress as a pathogenic driver.
The ER and the mitochondrial network are highly interconnected at the mitochondria-associated ER membrane (MAM). MAM disruption leads to an imbalanced Ca2+ flux into the mitochondria affecting energy production and leading to lower ATP content, and it has been found in models with mutations in SOD1, TARDBP, FUS, and VAPB. Promoting stability of the MAM, administering ligands of the Sigma1-receptor has shown therapeutic benefit in ALS mice. 137,138
Protein aggregates or ubiquitin-positive inclusions involving TDP-43, SOD1, FUS, ubiquilin 2 (UBQLN2), neurofilaments, and C9ORF72-related DRPs are well-established pathological hallmarks of ALS. While their role as primary toxic species is debated, high-molecular-weight complexes preceding aggregate formation are considered likely contributors to toxicity, potentially via prion-like intercellular propagation mechanisms.
TDP-43 cytoplasmic inclusions are present in neuronal and non-neuronal cells of patients with ALS, excluding those linked to SOD1 and FUS mutations. In contrast, wild-type TDP-43 is predominantly localized in the nucleus where it plays essential roles in RNA processing, including transcriptional regulation, alternative splicing, and miRNA processing. Other ALS-linked genes involved in RNA metabolism include FUS, EWSR1, HNRNPA1, HNRNPA2B1, and TIA1, which are known to form stress granules and impact RNA transport and splicing. In C9ORF72 ALS, repeat containing RNA foci sequestering RNA-binding proteins and repeat-associated non-AUG translation further perturb RNA metabolism and protein homeostasis.
The accumulation of misfolded proteins elicits the ER stress response, activating the unfolded-protein response (UPR), initially protective but becoming a maladaptive prodegenerative program under sustained stress. UPR markers are upregulated in patients with ALS as well as in mutant SOD1 mice. Interestingly, upregulation of several UPR markers occurs before muscle denervation in vulnerable large motor units but not in resistant ones, suggesting a role for ER stress in determining MNs’ susceptibility to degeneration.
Axonal transport defects also contribute to pathology. Different genes associated with familial ALS, such as PFN1, DCTN, TUBA4A, KIF5A, and NEFH, are involved in axonal transport. Several works have demonstrated accumulation of neurofilaments in MN and abnormalities of organelle axonal trafficking in patients with ALS, suggesting impaired cytoskeletal dynamics.
Finally, neuroinflammation is a consistent pathological ALS feature and has been proposed as a therapeutic target. MN degeneration initially leads to the activation of microglia, astrocytes, and the complement system, further contributing to the disease progression. Spinal cord and CSF from ALS cases present increased microglial activation and T cell infiltration, as well as higher concentration of proinflammatory mediators. Glial cells appear to play a crucial role in MN degeneration, as selective suppression of mSOD1 in MNs delays disease onset without affecting progression, whereas deletion in microglia and macrophages slows disease progression without altering onset. Complex signaling between CNS-resident immune cells and peripheral cells, including monocytes and T cells, has been reported. Thus, macrophages infiltrate peripheral nerves before spinal cord microgliosis in ALS mice. Astrocytes are likewise implicated in ALS pathogenesis. Astrocytes derived from both familial and sporadic patients with ALS are toxic to MNs in vitro, and blocking mSOD1 expression in these cells exerts neuroprotective effects.
GENE SUPPLEMENTATION STRATEGIES
The delivery of exogenous genes and their supraphysiological expression has been mainly centered on enhancing the production of neuronal survival and neurotrophic factors (NTFs), as well as modulators of NMJs. In contrast, alternative strategies aiming to prevent apoptosis, impede protein aggregation, or promote glutamate clearance have been studied to mitigate the insidious disease progression and the selective MN degeneration. These approaches are independent of the etiology of the disease; however, they are unlikely to provide a definitive cure, but rather they hold therapeutic potential by preventing motor function loss and extending survival.
Neurotrophic factors
Neurotrophic factors are small proteins that support the growth, survival, synaptic plasticity, and differentiation of neurons. A variety of NTFs have been shown to promote survival and axonal regeneration of damaged neurons in vitro and in animal models. The concentration of certain NTFs and growth hormones is altered in the CSF and blood of patients with ALS, as well as in ALS animal models. 139 Supplementation with these factors as recombinant proteins can support the ailing neuromuscular function, but inadequate dosing and delivery have led to unsuccessful therapeutics with important side effects. 140 Viral-mediated delivery can finely and precisely express NTFs in the desired tissues, avoiding constant readministration.
Glial cell line-derived neurotrophic factor (GDNF) is one of the most potent neurotrophins (NT), and its delivery through different routes and targeting strategies has been experimentally assessed in several studies. In transgenic SOD1 G93A murine models, overexpression of GDNF in the spinal cord has been tested, either through systemic administration of AAV9 vectors, 14,141 through i.s. transplantation of LV-modified stem cells, 12,15,17 or direct injection of LV vectors. 11 Despite robust GDNF expression in these studies, the resulting MN preservation and improvement in motor function were modest or even negligible in LV-GDNF-NSC-transplanted human patients (NCT02943850). 142 Moreover, whole-body overexpression in rats following systemic AAV9 delivery was associated with slower weight gain and reduction in activity and working memory, indicating that systemic administration of some growth factors may lead to adverse effects. To minimize such side effects and with the aim to promote axon guidance and GDNF retrograde transport, several groups opted for i.m. injections of AAVs, Ad, or ex vivo-modified MSCs in mSOD1 murine models. When administered into a single muscle, these approaches failed to produce clinically meaningful benefits, despite the preservation of MNs innervating the targeted muscle. 143 On the contrary, simultaneous administration into multiple limb muscles or combined with intercostal or paraspinal muscles delayed disease onset and extended survival by an average of 2 weeks. 6,8,13,144 Building on this concept, more recently we achieved widespread GDNF overexpression across all skeletal muscles through systemic administration of AAV8 vectors, using the muscle-specific desmin promoter to restrict expression to muscle tissue. This approach effectively preserved MNs, NMJs, and the compound muscle action potential (CMAP) amplitudes, resulting in delayed disease onset and slowed disease progression. 16
By applying analogous methods, insulin-like growth factor 1 (IGF-1) was evaluated through i.m. injections into various muscles of SOD1 G93A rodents using AAV or LV vectors. One particularly interesting study used AAV2 to retrogradely target MNs, resulting in a substantial 37-day extension of lifespan and a 31-day delay in the onset of motor deficits. However, the therapeutic benefits were markedly reduced when treatment was initiated at symptoms onset. 10 Subsequent studies employing alternative AAV serotypes failed to replicate these robust outcomes to the same extent but consistently demonstrated that IGF-1 contributed to the preservation of MNs, improved gait and rotarod performance, and modestly extended survival. 21,23,24 Similar findings were replicated with IGF-2—which signals through the same pathway—delivered i.m. with AAV9 vectors, 25 further supporting the neuroprotective role of the IGF1-PI3K-Akt pathway in MN maintenance and axonal regeneration. When focusing directly on MNs, i.s. injections of AAV2 resulted in the preservation of this particular cell type only within the treated spinal segments. 20 Notably, broader therapeutic benefits, such as delayed disease onset, slowed progression, and prolonged survival, were observed when IGF-1 was delivered i.s. to the lumbar spinal cord via AAV2 18 or systemically through i.v. administration of self-complementary (sc) AAV9 vectors. 22
The protective effects of IGF factors have been postulated to be partly dependent on the vascular endothelial growth factor (VEGF) signaling. Azzouz and collaborators were the first to demonstrate the therapeutic potential of VEGF on ALS, using retrogradely transported LV vectors encoding VEGF injected into various muscles of SOD1 G93A mice. 29 This strategy resulted in a 38-day extension of lifespan and a 28-day delay in the disease onset, comparable to the outcomes reported by Kaspar et al. with IGF-1. In a different model, AAV-VEGF vectors were injected i.c.m., i.v., or i.c. into the motor cortex of limb-expression 1-deficient feline models of lower MND. Although i.c. AAV1 injection confirmed anterograde transport along the corticospinal pathway, VEGF expression in the spinal cord remained low, similar to the levels observed following i.v. scAAV9. On the contrary, i.c.m. administration of AAV1 and scAAV9 vectors yielded robust transduction of the entire spinal cord, yet no therapeutic clinical benefit was detectable by any of these strategies. 30 Conversely, in the SOD1 G93A mouse model, i.t. administration of scAAV9-VEGF fostered survival and motor performance, 31,32 but to a much lesser extent than that observed with i.m. LV-VEGF vectors. Interestingly, i.c.v. coadministration of IGF-1 and VEGF delayed motor decline and extended survival although not in an additive manner, further emphasizing the possibility that these NTFs may act through overlapping signaling pathways. 53
Hepatocyte growth factor (HGF) is a multifunctional NT that has also been delivered i.t. and i.m. to SOD1 G93A mice using AAV vectors. Contrary to other growth factors, i.m. injections at three bilateral sites and retrograde transport of AAV6-HGF showed no effects on muscle strength and only modest signs of improvement in motor symptoms and survival. 26 This limited efficacy may be attributed to the substantially lower AAV doses in this study, 10- to 100-fold less per muscle compared with similar strategies. In contrast, i.t. administration of AAV1-HGF was reported to enhance rotarod, hanging wire and grip strength tests, and modestly extended lifespan. 27 More recently, the same authors investigated the underlying mechanisms of this treatment, demonstrating that i.t.-delivered AAV9-HGF attenuates gliosis, reduces ubiquitin accumulation, prevents vacuolization of MN apical dendrites, and mitigates upper MN loss in the motor cortex of prpTDP-43 A315T mice. 28 This supports the capacity of i.t. HGF delivery to modulate cortical pathology. Similarly, the i.s. administration of AAV1 encoding granulocyte-colony stimulating factor (G-CSF) to SOD1 G93A mice boosted muscle strength, delayed paresis, and increased survival by 10%, whereas i.m. delivery failed to transduce or preserve MNs. 36 Other NTFs—such as NT-3, cardiotrophin-1, and ciliary neurotrophic factor—have been less extensively studied, but they demonstrated comparable therapeutic outcomes in ALS mouse models when overexpressed in muscle. However, they were delivered using Ad vectors and administered postnatally. 33 –35
Multifactorial gene therapies for ALS have also been pursued, aiming for a more effective and holistic approach. When a combination of LV-modified MPCs populations expressing GDNF, VEGF, IGF-1, or BDNF was i.m. transplanted only into the gastrocnemius muscle (GM) muscle of SOD1G93A mice, it resulted in a significant preservation of both NMJ and MN axons. This approach delayed disease onset and extended survival by 10 days. 54 A similar approach involving a lower number of GDNF- and VEGF-MPC cells injected to the tibialis anterior, triceps brachii, and longissimus thoracis of SOD1G93A rats demonstrated the preservation of MNs and NMJs as well, delaying motor decline and extending survival by up to 28 days. 55
Modulators of the NMJ
Modulation of the NMJ has emerged as a promising gene therapy strategy for ALS. This approach is based on the “dying-back” hypothesis that maintains that the progressive death of MNs in ALS is preceded by failure and detachment of NMJs and axonal retraction, depending upon defects in the interaction of motor axons with terminal Schwann cells and skeletal muscle fibers. Thus, therapeutic strategies have been developed attempting to maintain the functional contact of the NMJ.
Several studies have implicated Neuregulin 1 (Nrg1) isoforms in neuromuscular health, with isoform I (Nrg1-I) contributing to the development and maintenance of the NMJ and isoform III (Nrg1-III) playing a role in MN preservation. 145 Loss-of-function mutations in ErbB4—a receptor for Nrg1—have been linked to a genetic form of ALS. 146 In a previous study, we demonstrated that i.m. injection of AAV1-Nrg1-I into the GM enhanced NMJ innervation and increased CMAP amplitude via collateral sprouting, without conferring protection to axons or MNs. 38 Notably, only systemic AAV8-mediated Nrg1-I overexpression across all skeletal muscles preserved MNs, neuromuscular function, and locomotor performance, ultimately delaying disease onset. 40 The i.t. infusion of AAV9-Nrg1-III produced similar benefits in female mice, 41 whereas the i.s. delivery of AAV1-Nrg1-III extended lifespan in both sexes. 39 Noteworthily, combining AAV8-Nrg1-1 and AAV9-Nrg1-III along with dual-route administration (i.v. and i.t., respectively) failed to yield additive or synergistic effects. 147
In an alternative strategy, docking protein 7 (DOK7), essential for NMJ formation, was ubiquitously expressed following AAV9 tail vein delivery. While DOK7 did not prevent MN loss, it protected from nerve terminal and muscle atrophy, clustering AChR and enlarging NMJs, resulting in a modest increase in lifespan (7.5%) and motor activity. 42 More recently, Chen et al. overexpressed nuclear receptor interaction protein (NRIP) in the skeletal muscles of SOD1 G93A mice. 43 NRIP, critical for sarcomere integrity and NMJ stability through AChR binding, is markedly downregulated in SOD1 G93A spinal cord and muscle, and its loss results in progressive MN degeneration. The i.m. AAVDJ/8-NRIP delivery to fore- and hindlimb muscles improved locomotor activity, enlarged myofibers, reduced NMJ and axonal degeneration, and preserved MNs and CMAPs but did not sustain grip strength or extend survival.
Neuroprotective strategies
Other researchers have focused on preventing MN death rather than promoting their survival. Azzouz et al. exemplified this approach by delivering bilateral i.s. injections of AAV2 into the lumbar spinal cord to overexpress the antiapoptotic protein Bcl-2. 44 This strategy rescued nearly 50% of the MNs, preserved CMAP amplitude, and successfully delayed disease onset. However, local Bcl-2 expression alone was insufficient to extend overall survival. In a more recent investigation, muscle overexpression of synaptotagmin 13 (SYT13)—a vesicular trafficking protein enriched in extraocular MNs and important for synapsis and vesicle metabolism—was tested in both ALS and SMA mouse models. Gene therapy with SYT13 led to a 14% extension in lifespan in ALS mice and a remarkable 50% in the SMA model by decreasing MN apoptosis and mitigating muscle denervation. 45
Several studies have specifically investigated strategies aimed at reducing overall toxic protein aggregation in the spinal cord of ALS mice. Among these, single-chain fragment variable (scFv) antibodies have been used to directly target aggregated forms in different preclinical models of ALS, effectively preventing their propagation and toxic effects. 46,148 –150 Notably, the study by Patel et al. delivered scAAV1 vectors to achieve sustained expression of scFv antibodies against mSOD1 in the spinal cords of presymptomatic adult SOD1 G93A mice, resulting in a significant 28% extension in survival and delayed disease onset. 46 Since this treatment targets the underlying cause of the disease in this model, its relevance is therefore narrowed to mutations in the SOD1 gene. A related strategy involved the delivery of serine-rich chaperone protein 1 to the motor cortex via AAV9 vectors, which resulted in only modest reductions in SOD1 aggregates and failed to produce any survival benefit, highlighting the limitations of this chaperone and/or a suboptimal CNS coverage. 49
Another protein with chaperone-like properties, macrophage migration inhibitory factor (MIF), has also shown promise. MIF expression is reduced in human induced pluripotent stem cells (iPSC)-derived MNs from familial patients with ALS, as well as in postmortem tissues from individuals with sporadic ALS. In an initial study, MIF was delivered via AAV2 vectors i.s. to postnatal day 0 (P0) SOD1 G93A mice, resulting in a significant delay in disease onset and a 20% extension in survival. 47,151 A subsequent study used the blood–brain barrier (BBB)-penetrant AAV-PHP.eB vector to achieve widespread CNS delivery of MIF in SOD1 G37R mice. 48 This approach reduced neuroinflammation, rescued MNs, and corrected dysregulated pathways, as shown through omics analyses. However, in this case, survival extension was more modest (6.8%), likely due to administration at symptoms onset. While this approach is more clinically relevant, it may limit capacity to clear pre-existing mSOD1 aggregates. Further research would elucidate whether this strategy can be applied to other ALS models characterized by the accumulation of misfolded proteins.
Increasing autophagy through enhanced expression of optineurin (OPTN) has been proposed as a potential therapeutic strategy in ALS. Mutations in the OPTN gene have been reported in patients with ALS, associated with low autophagic activity. OPTN plays a role in mitophagy and in the recruitment of ubiquitinated substrates for autophagosome-mediated degradation. In ALS, however, OPTN colocalizes with ubiquitin and TDP-43-positive inclusions, suggesting sequestration and dysregulation of its function. In SOD1 G93A mice, OPTN is increased during the presymptomatic phase but declines after disease onset. 64 Two recent studies overexpressed OPTN in the spinal cord of SOD1 G93A mice. One study delivered AAV9 vectors i.t. at a presymptomatic age, improving stride length, rotarod performance, and extending lifespan by 10.5%. 63 The second study delivered OPTN using LV vectors administered into the right lateral ventricle. Presymptomatic LV-OPTN treatment postponed disease onset and resulted in only a 7.2% increase in survival, despite increasing by 46.8% the preservation of spinal MNs at disease onset. Notably, symptomatic administration failed to produce significant effects on disease progression or survival. 64 These findings indicate that OPTN expression may be beneficial before disease onset and were attributed to enhanced autophagic and mitophagic flux, preserved mitochondrial morphology, anti-inflammatory responses, and reduced apoptosis.
Additional gene therapy strategies have aimed to counteract glutamate excitotoxicity, a key pathogenic mechanism in ALS. DAO, EAAT2, or adenosine deaminase RNA-specific B1 (ADAR2) were overexpressed with the help of AAV vectors in the CNS. 50 –52 DAO metabolizes d-serine, a coagonist of NMDA receptors involved in excitatory neurotransmission; EAAT2 is exclusively expressed in astrocytes and serves as the primary transporter responsible for clearing extracellular glutamate; and ADAR2 is essential for editing the Q/R site of GluA2 mRNA, which determines the calcium permeability of AMPA receptors. Despite the rationale behind these antiglutamatergic strategies, none resulted in significant improvements in motor strength or locomotor function, and only DAO provided a modest extension of survival in mSOD1 mice.
Using a less conventional strategy, Benkler and coauthors delivered a multifactorial cocktail i.c.m. to the CNS and i.m. to the GM muscles to address the excitotoxic and oxidative axes. 56 The mixture contained LV vectors carrying EAAT2, glutamate-dehydrogenase 2 (GDH2), and nuclear factor-related factor 2 (NRF2) genes. While EAAT2 is related to glutamate uptake, GDH2 metabolizes glutamate and reduces its bioavailability. NRF2, a vital regulator of antioxidant and anti-inflammatory pathways, was selected to address oxidative stress. This combination produced notably remarkable outcomes, as all three genes together but not separately prolonged survival in SOD1 G93A mice by an average of 19–22 days. This was accompanied by improvements in all neurological and motor parameters evaluated.
Collectively, gene overexpression therapies targeting skeletal muscles in mSOD1 models have generally yielded more favorable outcomes than CNS-directed strategies, particularly when delivering NTs or using retrogradely transported viral vectors. Furthermore, some of the strategies offered cumulative effects with combinatory treatments, underscoring the significance of synergistic, multitargeted approaches in addressing the complex pathophysiology of ALS.
GENE SILENCING STRATEGIES
SOD1 silencing
A rational approach for treating ALS involves silencing genes that contribute to the disease through a gain-of-function mutation, particularly those responsible for familial forms of ALS. As the first-identified gene linked to ALS, SOD1 has been silenced by multiple studies. In this context, the paradigm shifts toward evaluating and prioritizing the degree of silencing and survival as primary outcomes. The first two studies, concurrently published, delivered RNAi therapy against SOD1 in SOD1 G93A mice using LV vectors. The study with more favorable outcomes targeted multiple muscles (face, tongue, diaphragm, hindlimb, and intercostal muscles) at a postnatal stage using LV based on the equine infectious anemia virus (EIAV), which is known to undergo retrograde transport following i.m. injection, resulting in up to a 77% increase in survival and a delay in disease onset of more than 100%. 65 The other study focused on bilateral i.s. injections to the lumbar spinal cord of adult mice before the onset of motor dysfunction. Although it did not significantly extend survival, it retarded onset and slowed disease progression. 66
In line with the importance of vector tropism and transport properties, Miller et al. investigated muscle-specific toxicity by comparing AAV1 and murine LV vectors in SOD1 G93A mice. 67 As murine LV lacks retrograde transport capacity, i.m. injection restricted siRNA expression to muscle, whereas AAV1 enabled silencing in both muscle and spinal motor neurons. Only the latter improved grip strength and muscle mass. Furthermore, Cre-mediated knockdown of floxed mSOD1 G37R specifically in muscle had no impact on functional outcomes. Similarly, AAV-mediated follistatin overexpression enhanced muscle mass and strength but did not affect onset or survival, suggesting that muscle is not a major contributor to noncell-autonomous toxicity in this model.
As the i.s. injection into a single or few spots can only cover a small fraction of MNs, a different study directly infused modified small interfering RNAs (siRNAs) intrathecally at the symptoms onset using a pump-based delivery system. siRNAs efficiently knocked down SOD1, slowed down the progression, and slightly extended survival. 68 Interestingly, Wu and colleagues attempted to reach MNs by an intrasciatic nerve injection in an aim to cover more MN pools and use lower viral doses. 75 They demonstrated that Ad-U6-shSOD1 targeted lumbar MNs more efficiently than AAV2, resulting in a deceleration of body weight loss and a 6% increase in survival.
Subsequent studies have focused exclusively on AAV-mediated delivery of shRNA. Unexpectedly, and contrasting to results obtained with EIAV-LV vectors, injection of AAV6-H1-shSOD1 muscles across the body of SOD1 G93A mice failed to improve motor performance or extend survival. 74 It is unlikely that the discrepancy in therapeutic outcomes can be solely attributed to differences in MNs transduction efficiency, as the initial LV study reported transduction in over 50% of MNs versus 40% in the AAV study. Conducting a thorough characterization of the lower MN axis following vector transduction would help to determine whether a threshold of MN transduction is required for therapeutic benefits via i.m. delivery and retrograde transport.
Initially, i.v. delivery of AAV-shSOD1 vectors conferred no clinical benefits, 73 probably attributed to the use of the AAV6 serotype, which does not cross the BBB efficiently. Conversely, i.v. administration of slightly higher doses of AAV9-shSOD1 improved motor function, retained muscle mass, and significantly extended survival in SOD1 transgenic mice. This effect was age-dependent, ranging from 22% to 39% across the postnatal age to the diseased age spectrum. Interestingly, only mice treated at P1 exhibited delayed disease onset due to more effective MN SOD1 silencing, whereas those treated at P21 displayed slower disease progression, linked to enhanced transduction of non-neuronal cells. 72 Intracisterna magna or intracortical delivery of AAV9-shSOD1 resulted in a less effective correction of motor symptoms, disease onset, and survival (12–13%). 69,71 Finally, a more recent study described a novel subpial injection technique for smoothly delivering AAV9 through all the spinal cord and brain motor centers of mice, pigs, and nonhuman primates (NHPs). Among all SOD1-silencing strategies reported to date, this study has shown the most promising outcomes. The authors successfully suppressed MND, achieved near complete preservation of MNs and muscle innervation, with a full correction of survival, and a block in progression when administered after symptoms onset. 70
miRNAs are part of the cell natural regulatory machinery and, rather than completely silencing, can partly reduce mRNA levels. In comparison with shRNA, they offer a more precise targeting, endogenous regulation, and lower immunogenicity and can be combined with specific promoters, all lowering off-target effects. 152 The intra-CSF AAV-miRSOD1 delivery at postnatal stages to SOD1 G93A mice sustains survival to varying degrees, depending on the serotype and dose administered. 82,83,86 At that age, specifically targeting astrocytes extended survival and rescued neuromuscular function, although not to the same extent as when targeting MNs, and it did not result in a delay in the disease onset. 83 Conversely, i.t. injections in adult nonsymptomatic mice induced SOD1 silencing and enhanced locomotor function but provided scarce to no extension of lifespan. 79 –81,83 A phase I/II clinical trial is currently ongoing using AAVrh10 delivered i.t. (NCT06100276).
ASOs have been thoroughly investigated in neurodegenerative diseases and even progressed to clinical trials. Typically delivered without vectors, they offer advantages including an improved safety profile, easy administration, high tissue penetration, and rapid onset of action. The first study evaluating ASO therapy in SOD1G93A rats employed a modified ASO targeting SOD1, continuously infused intraventricularly. This approach demonstrated the feasibility of reducing SOD1 mRNA and protein levels in both the spinal cord and brain. Although treatment was initiated near disease onset and did not delay it, it significantly slowed disease progression and modestly extended survival by 8%. 91 These findings led to the first-in-human phase I clinical trial of i.t. ASO delivery, which reported no dose-limiting toxicities, tolerability of redosing and validated the i.t. route for CSF administrations (NCT01041222). 92 Subsequent studies in SOD1 G93A rats using next-generation ASOs at higher doses, administered via i.c.v. or i.t. routes, showed improved outcomes. These included increased survival rates (22% and 32%−39%), delayed disease onset (by 30.7% and 14.8%), improved locomotor function, and reversal of the initial CMAP decline. 96 This paved the way for the next-generation compound, BIIB067 (Tofersen), which was evaluated in a follow-up phase I/II clinical trial (NCT02623699) involving repeated i.t. dosing at 20–100 mg over a 12-week period via lumbar injections. The trial established the safety of higher dosing regimens and demonstrated a 33% reduction in CSF SOD1 protein levels in the highest-dose cohort, along with preliminary evidence of slowed functional decline. 93 Most recently, the phase III trial concluded, showcasing both reduced CSF SOD1 concentrations and neurofilament light chain (NFL) plasma levels. Although the primary clinical endpoints were not met and the therapy showed adverse events, Tofersen was approved by regulatory agencies as an investigational drug under the name Qalsody. 94 The trial included an open-label extension phase (NCT03070119), enabling a delayed-start comparison between participants who began Tofersen at trial entry and those who switched from placebo after 28 weeks. This phase also supported long-term evaluation of BIIB067, providing data for up to 1 year. Results suggested a slower rate of functional decline in those who initiated therapy earlier. However, clinical interpretations are limited by the unblinded and uncontrolled design of the extension phase. Given these results, a confirmatory phase III trial (ATLAS, NCT04856982) is currently ongoing in presymptomatic SOD1 mutation carriers, aiming to determine whether early initiation of Tofersen can delay disease onset and progression.
In parallel to ASO-based strategies, a gene therapy approach using AAVrh10 vectors has also shown strong efficacy in the SOD1 G93A mouse model. This approach mediated exon skipping of the SOD1 pre-mRNA by delivering exon 2-targeted antisense sequences embedded in a modified U7 small nuclear RNA (AAV10-U7-hSOD1). Exon 2 skipping resulted in the introduction of a premature termination codon, effectively disrupting SOD1 protein production. A single administration of AAV10-U7-hSOD1 via combined i.c.v. and i.v. routes at P1 resulted in a notable 92% extension in survival and a substantial 90% delay in onset, whereas treatment at P50 still led to a significant 58% increase in survival and a 76.5% delay in onset. 95 These findings highlight the strong therapeutic potential of early, systemic gene therapy targeting SOD1 expression as a powerful alternative or complementary approach to ASO-based interventions.
The emerging field of genome editing has also been explored in the context of ALS. The first clustered regularly interspaced short palindromic repeats (CRISPR)-mediated editing of SOD1 and delivery of Staphylococcus aureus-derived Cas9 (SaCas9) and single-guide (gRNA) against SOD1 was published in 2017. 107 Gaj and collaborators used the modified AAV9-2YF vector administered via the facial vein in SOD1 G93A neonatal pups. The approach yielded a ≥2.5-fold decrease in SOD1 levels in the spinal cord, resulting in improved motor function and reduced muscle atrophy. CRISPR-edited mice had 50% more surviving MNs at end stage, displayed a 37% delay in disease onset, and a 25% increase in survival. A subsequent similar study using i.c.v. delivery of AAV9-SaCas9-sgRNA at a lower dose in neonatal mice achieved comparable results, including a 54.6% increase in lifespan. 108 More recently, the BBB-penetrating AAV-PHP.B and PHP.eB vectors encoding a sgRNA were tested for SOD1 silencing in bigenic mice harboring the human SOD1 G93A and Cas9 transgenes. The i.c.v. administration of AAV-PHP.B immediately after birth reduced spinal MNs loss, denervation of NMJ, and muscle atrophy. This therapy also diminished axonal damage and preserved CMAP through animals’ lifespan, which was extended between 66% and 85% depending on the cohort. Importantly, i.v. or i.t. injections of AAV-PHP.eB in young adult mice immediately before symptom onset fully preserved neuromuscular and motor function, correcting survival (by ≥200%) at least until all animals had to be euthanized due to ethical endpoints rather than disease progression. The treatment in adult mice rendered a higher degree of mSOD1 protein reduction and stronger therapeutic benefits and may reflect the superior CNS transduction efficiency of AAV.PHP.eB versus AAV.PHP.B vectors as well as differences in the route of administration. 109
Finally, in vivo base editing, which enables single-nucleotide changes without introducing double-strand breaks, was explored in a study via i.t. injection of dual AAV particles encoding a split-intein cytidine base editor engineered to introduce a nonsense mutation into the mSOD1 gene. Treated adult SOD1 G93A animals had a prolonged survival (11%) and a marked slowed disease progression, with a reduced rate of muscle atrophy and denervation, improved neuromuscular function, and up to 40% fewer SOD1 immunoreactive inclusions. 111 This approach offers high target specificity while preserving genomic integrity. However, its overall efficiency is limited by the need of successful cotransduction of target cells with both AAV vectors, followed by effective trans-splicing to reconstitute the functional base editor.
C9ORF72 silencing
Silencing strategies for C9ORF72-linked ALS have been more challenging compared with SOD1-related ALS. The pathogenic expansion in C9ORF72 involves a long and complex hexanucleotide repeat in contrast to the point mutations observed in SOD1 ALS. Identifying optimal target sites is laborious due to its repetitive nature, and neutralizing the toxic RNA without compromising the normal C9ORF72 gene function poses a significant difficulty. Furthermore, C9ORF72 ALS exhibits a high degree of genetic and phenotypic heterogeneity, complicating the development of a one-size-fits-all silencing therapy. 153 Tailoring therapies for different variants and disease manifestations is a more intricate task, which adds to the limitation of animal models that do not accurately replicate this disease phenotype.
The first study investigating C9ORF72 silencing was conducted on patient-derived fibroblasts and demonstrated that effective reduction of nuclear RNA foci requires targeting both the sense and antisense strand repeat-containing RNAs with ASOs, as abundant antisense RNA foci (GGCCCC) were also observed. In contrast, siRNAs failed to reduce nuclear RNA foci despite markedly reducing overall C9ORF72 RNA levels, consistent with the cytoplasmic localization of the RISC complex and the primary site of siRNA activity. The i.c.v. administration of ASOs in wild-type mice reduced 30–40% the mRNA levels of C9orf72, without inducing neuropathological or behavioral abnormalities. 97 In a subsequent study, sense-targeting ASOs were administered i.c.v to C9ORF72 BAC transgenic mice carrying 450 repeats, achieving a 20–40% reduction in the cortex and spinal cord, while sparing wild-type C9orf72 mRNA levels. This treatment also reduced DRP inclusions and ameliorated age-related anxiety and cognitive deficits when administered at later stages of disease progression. 98 These promising results paved the way for advancing this treatment (BIIB078) into a phase I/II clinical trial (NCT03626012).
A separate study by Tran et al. provided the first clinical evidence of successful C9ORF72 suppression using a chemically modified ASO (afinersen) targeting the intronic region surrounding the repeat expansion. The study demonstrated target engagement and polyGP reduction in the CSF following repeated i.t. administration in a single human subject harboring approximately 2,400 G4C2 repeats. The patient’s ALS Functional Rating Scale score remained largely stable, with no meaningful clinical improvement or decline. To date, no clinical trial has yet been initiated based on this ASO program. 100
In another study, a different C9ORF72 BAC transgenic mouse harboring 800 G4C2 repeats was used to evaluate the efficacy of an experimental stereopure ASO (WVE-004). This ASO selectively and dose-dependently reduced sense repeat-containing transcripts, achieving CNS reductions of 59–84% following multiple i.c.v. administrations and 65% in iPSC-derived MN. The effects were long-lasting, persistently reducing transcripts and polyGP DPRs for at least 6 months. 99 As a result, WVE-004 progressed to clinical evaluation for patients with the C9ORF72 G4C2 expansion (NCT04931862). Despite encouraging preclinical results, both clinical trials, BIIB078 and WVE-004, failed to show clinical benefits and were associated with elevated NFL levels. These outcomes have raised concerns about potential C9ORF72 loss-of-function, contributions from antisense repeat transcripts, DNA-level toxicity, and/or inadequate target engagement. 101,154
Suboptimal target engagement may be attributed by timing of therapeutic administration or low biodistribution to neurons, despite achieving approximately 50% reductions in polyGP levels. Clinical benefit could emerge with earlier treatment or longer dosing durations, as seen for the open-label extension of the Tofersen trial for SOD1-related ALS. 94 However, the observed increases in NFL levels in both investigational arms raise the possibility of ASO-associated neurotoxicity when delivered in the CSF. While current clinical data do not provide clear evidence of ASO-induced neurotoxicity in humans, some preclinical studies have reported neuronal toxicity in mice, characterized by seizure-like phenotypes and dose-dependent reductions in consciousness and locomotor function, potentially linked to disruptions in intracellular calcium homeostasis. 100,155 –157
Moreover, these studies primarily focused on polyGP and polyGA levels, while overlooking the most toxic, insoluble aggregates formed by polyGR or omitting the measure of antisense-derived DPRs. Some evidence suggests that ASOs targeting the sense intronic region may exert an inhibitory bystander effect on antisense RNA transcription. 158 –160 Thus, it would be valuable to assess patient samples from these clinical trials for the levels of polyGR and polyPR to better understand how the spectrum of DPR suppression may contribute to therapeutical efficacy.
In contrast, although C9orf72 knockout (KO) mice exhibit immune abnormalities, they do not develop MN degeneration or ALS-like motor deficits. However, suppression of the endogenous wild-type allele in G4C2 repeat-overexpressing mice impairs autophagy, alters microglial function, and exacerbates disease progression. These findings highlight that C9orf72 loss-of-function should be considered a relevant contributor to disease pathogenesis and a potential limitation in gene-silencing therapeutic strategies. 161 –163
In a different approach, Martier et al. delivered AAV5 vectors coding concatenated miRNA hairpins through intrastriatal (i.s.t.) injections to Tg(C9orf72_3) line 112 mice (BAC112), a strain not displaying neurodegeneration but rather RNA foci and polyGP proteins. 87 They achieved a 20–40% reduction in C9orf72 mRNA and 20% fewer cells with RNA foci. Another study also targeted the striatum of BAC112 mice with artificial miRNAs delivered via AAV9 vectors, resulting in approximately 50% reduction in total C9orf72 transcripts and polyGP DPRs. 88 However, despite the stable expression from viral vectors, their reliance on cytoplasmic RNA processing pathways may limit therapeutic potential, given that pathogenic repeat RNAs are predominantly localized in the nucleus.
The failure of ASO clinical trials highlights the need for therapeutic strategies that can target C9ORF72 antisense repeat RNA transcripts and intervene at the DNA level. Genome editing approaches aimed at suppressing pathological C9orf72 expression have been investigated in only a few in vivo studies, most lacking functional outcome analyses. A first study demonstrated that delivery of AAV9-mediated CRISPR/Cas9 could excise the hexanucleotide repeat expansion from its genomic locus using gRNA flanking the repeat in three different C9BAC models (BAC111/Cas9, BAC112, and C9-500 mice). 112 More recently, two back-to-back studies employed a high-fidelity CRISPR-Cas13 system, which cleaves RNA through its intrinsic RNase activity—offering a reversible and potentially safer approach for modulating gene expression—and is compact enough for packaging into AAV vectors. 113,114 Both studies targeted sequences upstream of the G4C2 repeat to minimize off-target effects associated with the repeat’s high GC content and its presence elsewhere in the genome. This strategy enabled selective targeting of C9orf72 transcript variants 1 and 3, while sparing variant 2, the predominant isoform in the brain, which initiates transcription downstream of the repeat, thereby reducing the risk of C9orf72 loss of function. Notably, the study by Kempthorne et al. leveraged CasRx variant ability to process multiple guide RNAs from a single construct, enabling concurrent reduction of sense and antisense transcripts. Their approach was validated in two different mouse models and also demonstrated neuroprotection against glutamate-induced excitotoxicity in iPSC-derived MNs. 113
FUS silencing
In an initial proof-of-concept study, the FUS gene was suppressed in the common marmoset by delivering shRNA through AAV9, directly injected into the frontal cortex. This approach resulted in robust FUS silencing (approximately 70–80%) across various cell types, provoking an increased proliferation of astrocytes and microglia. 76 However, no additional shRNA-based approaches have been reported to date.
More recently, ASOs against FUS were introduced i.c.v. in newborn FUS knock-in MN-P517L/Δ14 mice, demonstrating protective effects against MN loss and denervation for up to 4 months. 102 As ASO levels gradually decreased, microgliosis intensified but without subsequent MN degeneration. This ASO (ION363 or Ulefnersen) obtained the Food and Drug Administration’s approval for experimental compassionate use in a young woman reliant on ventilatory support. Administered i.t. and initiated 6 months after clinical onset, the ALS-FRS-R score decline rate slowed during the treatment course. 102 Unfortunately, the patient’s condition later deteriorated, particularly in ventilatory and bulbar function, and ultimately passed away due to complications. The therapy, named jacifusen in her honor, is currently undergoing recruitment in a phase III clinical trial (NCT04768972). Previously, a series of single-patient investigational new drug applications has suggested the safety and possible efficacy of jacifusen for treating FUS-ALS. 103
Targeting TDP-43 pathology
TDP-43 aggregation is a pathological hallmark of nearly all ALS cases; however, effective therapies directly targeting TDP-43 for ALS/FTD and related TDP-43 proteinopathies remain elusive. Given the essential role of TDP-43 in RNA metabolism and cellular homeostasis, direct silencing of TDP-43 may be unsuitable as a therapeutic strategy. As an alternative, Becker et al. proposed targeting ataxin-2 (ATXN2) to disrupt TDP-43 aggregation. 104 Intermediate-length trinucleotide repeat expansions in the ATXN2 gene are a known genetic risk factor for familial ALS and have been implicated in stress granule formation, promoting aberrant TDP-43 cleavage and its mislocalization to stress granules. 164 –166 In a proof-of-concept study, newborn TAR4/4 mouse pups were treated i.c.v with ASOs against Atxn2, resulting in a 35% extended lifespan in this severely affected model, which typically reaches humane endpoints around day 21. Beyond prolonging survival, reducing Atxn2 improved gait scores and slowed disease progression. These data supported the advancement of this ASO, BIIB105 (also known as ION541), into a phase I/II clinical trial (NCT04494256). The trial involved i.t. administration to patients with ALS with or without ATXN2 repeat expansions. However, the study was recently terminated after reporting no reduction in NFL levels or improvement in functional, respiratory, or strength-related outcomes over a follow-up period of more than 40 weeks, despite a reduction in ATXN2 protein levels in CSF.
In a separate study, a Cas13-based gene silencing approach targeting Atxn2 system was delivered to neonatal TAR4/4 pups via AAV9 administered through combined i.c.v. and i.v. routes. 115 This approach led to significant therapeutic benefits, including improved gait, reduced kyphosis and tremors, increased body weight, and a 137% increase in survival, extending lifespan by 35 days. In the same mouse model, AAV-mediated RNAi delivery using a novel capsid (PM-AAV9) administered i.c.v. at P1 also extended survival, albeit to a lesser extent, consistent with its knockdown mechanism. 77 RNAi treatment resulted in a 45.5% increase in median survival, suggesting that while both approaches are beneficial, Cas13-mediated KO may confer a more robust therapeutic effect. To assess the therapeutic potential of Atxn2 reduction in a non-TDP-43 overexpressing ALS mouse, Hawley et al. developed a miRNA-based approach targeting Atxn2 in the PFN1 C17G model, which exhibits strong TDP-43 pathology. 90 Mice were injected i.t. with AAV-PHP.eB-mAtxn2-amiR at 6–9 weeks of age, coinciding with the emergence of insoluble pTDP-43 accumulation. This intervention delayed the rise of serum NFL levels and was associated with preserved body weight gain, motor function, and muscle strength. However, effects on survival were not assessed.
Directly targeted TDP-43 has also been assessed with ASO-based therapies. Takeuchi et al. employed gapmer-type ASOs incorporating 2′-O,4′-C-ethylene-bridged nucleic acids (ENAs) to induce a transient and specific reduction of human TDP-43 expression. These highly stable, ENA-modified ASOs were administered i.c.v. to presymptomatic 6-week-old mice. A single injection effectively lowered TDP-43 levels throughout the CNS, resulting in 12 weeks of sustained suppression of cytoplasmic TDP-43 aggregation and lasting improvement in behavioral and locomotor deficits, even after pathological TDP-43 levels returned. 105 However, the study did not assess potential toxicities from suppressing endogenous TDP-43, as mouse TDP-43 was not targeted by gapmer ASOs, a critical consideration for clinical translation.
Along a similar line, Ke et al. leveraged their discovery of a noncanonical interaction between TDP-43 and 14-3-3θ to specifically target the aberrant accumulation of TDP-43 without affecting the expression or function of endogenous mRNA. 117 The chaperone-like protein 14-3-3θ selectively binds to pathological TDP-43 variants, exacerbating the disease. They engineered a gene therapy construct, DD-θFx, which fuses a degron to the α6 helix of 14-3-3θ. This modification enables proteasomal degradation of misfolded TDP-43. AAV-mediated delivery of DD-θFx effectively reduced toxic TDP-43 accumulation in three ALS mouse models: TDP-43 A315T model, AAV-induced human TDP-43 overexpressing mice, and the aggressive rNLS8 model. In the TDP-43A315T and AAV-hTDP-43 models, DD-θFx improved behavioral and motor deficits. In the rNLS8 model, DD-θFx treatment significantly delayed paralysis onset and extended survival by 38.5%. This study underscores the therapeutic potential of disrupting the pathological 14-3-3θ/TDP-43 interaction as a targeted strategy to reduce TDP-43 aggregation and mitigate neurodegeneration in ALS.
TDP-43 loss drives disease through splicing dysregulation in two critical neuronal genes: STMN2 and UNC13A by reducing the expression of proteins essential for neuronal function. Loss of function in STMN2 leads to motor neuropathy and denervation of the NMJ in mice. 167 In patients with ALS, nuclear TDP-43 depletion causes addition of a premature polyA tail in STMN2, reducing its protein function. In cellular models and in humanized mice carrying the cryptic intronic sequence of STMN2, gene editing using dCasRx—a catalytically inactive Cas13 guided to the splice site—or i.t. administration of ASOs restored proper splicing. This correction led to normalized STMN2 mRNA and protein levels, improved axonal regeneration following injury, and rescued lysosomal trafficking. 106 Notably, the ANQUR phase I clinical trial (NCT05633459) is evaluating the ASO QRL-201 to restore STMN2 expression in patients with ALS lacking SOD1 and FUS mutations. In contrast, UNC13A is a gene essential for synaptic vesicle fusion. When TDP-43 is lost from the nucleus, a cryptic exon is included in the UNC13A transcript, introducing a premature stop codon and triggering degradation via nonsense-mediated decay. In patients with ALS, higher levels of cryptic splicing and more severe reduction of UNC13A expression are strongly associated with faster disease progression. 168,169 In a more sophisticated approach, Wilkins and colleagues developed a novel gene therapy platform called TDP-REG, which harnesses cytotoxic cryptic splicing induced by TDP-43 loss-of-function, to control therapeutic gene expression precisely in diseased cells, when the cryptic exon is inserted upstream of their constructs. They used a TDP-43/Raver1 fusion protein that acts as a splicing repressor, restoring normal splicing for UNC13A and STMN2 both in patients’ iPSC-derived MNs and in TDP-43 conditional KO mice. 116
CONCLUDING REMARKS AND FUTURE DIRECTIONS
Gene therapy has emerged as a promising approach for ALS, enabling molecularly targeted interventions in a disease with multifactorial origin and unmet medical needs. Preclinical studies—particularly in SOD1-related mouse models—have shown that AAV-based delivery of NTFs can delay disease onset, preserve MNs and NMJs, and moderately extend survival, especially when administered early. Multifactorial strategies combining neurotrophic, anti-inflammatory, and antioxidant genes have demonstrated additive effects while targeting NMJ stability and maintaining muscle–nerve connectivity. In contrast, muscle-targeted delivery can preserve NMJs and delay MN death by synaptic contacts. Moreover, combining peripheral and central approaches may enhance efficacy.
Gene silencing has demonstrated even greater therapeutic potential, particularly in the context of SOD1 mutations, achieving near-complete preservation of MNs and normalization of survival in preclinical models. As a result, ASO-based therapies have progressed to clinical trials, leading to the conditional approval of Qalsody for a small subset of patients. Despite this milestone, clinical outcomes have shown mixed efficacy across key endpoints. It remains critical to assess the therapeutic benefit of this treatment in a broader patient population, especially considering that, to date, clinical trials in ALS have largely failed to yield consistently positive results.
Although new therapeutic platforms have shown great promise for treating neurological diseases such as ALS, their safety profiles remain a key concern. In the case of AAV vectors, although the CNS is relatively immune-privileged and usually only a single administration is needed, high-dose i.v., or even i.t. or i.c.m. delivery has been associated with dorsal root ganglia toxicity, inflammation, and neuronal degeneration in both preclinical and clinical settings. 170 Moreover, pre-existing neutralizing antibodies against AAV capsids and immune responses to the transgene product can compromise efficacy and induce adverse effects. 171 To mitigate these risks, strategies include using lower doses, neuron-specific or endogenous promoters, immunosuppressive regimens, and novel capsids with enhanced CNS tropism and reduced immunogenicity. 172,173
ASOs are administered intrathecally to bypass the BBB. Their safety depends largely on the sequence, chemistry, and dosing regimen (usually weekly or monthly for the duration of the patient’s life). Some ASOs have been associated with neurotoxicity, motor dysfunction, or inflammatory responses in the spinal cord and brain in animal models, often influenced by the physiological importance of the wild-type function of the targeted protein. 174 However, Tofersen has shown acceptable safety profiles, with the most common side effects related to lumbar puncture. 175
Despite its revolutionary potential, CRISPR gene editing still poses significant safety concerns, especially in the context of permanent changes to the genome. One of the most critical risks is off-target editing, where the CRISPR-Cas system may cut DNA at unintended sites, potentially disrupting essential genes or activating oncogenes. 176,177 Moreover, delivery remains a critical factor: often AAV vectors are still necessary to deliver CRISPR tools into the CNS, leading to persistent expression of Cas9, increasing the window for genomic damage. Immunogenicity is another issue: the bacterial origin of Cas proteins can provoke immune responses that lead to inflammation or clearance of edited cells. 178 Alternative systems such as high-fidelity Cas9 variants, DNA break-free editing systems, as well as editing approaches that avoid double-strand breaks—such as base editors and prime editors—are being studied to increased CIRSPR gene editing. 179,180
Collectively, the reviewed data highlight the difficulty of treating this pathology, the importance of early intervention, and combinatorial targeting. Still, challenges remain—immunogenicity, delivery efficiency, and variability across ALS subtypes must be addressed. Translation to human therapy will require precise patient stratification, early biomarker development to identify the disease before its onset, and long-term studies.
Future directions include refining viral vectors, combining systemic and CNS-targeted approaches, using disease models beyond SOD1 mutants, including patient-derived and additional animal models, and expanding genome editing strategies. While no single treatment is expected to cure ALS, a synergistic, multitargeted approach may ultimately convert this devastating disease into a manageable chronic condition.
Footnotes
ACKNOWLEDGMENT
Figures were created with BioRender.com.
AUTHORS’ CONTRIBUTIONS
S.V. wrote the original draft of the article and created the figures. X.N. and A.B. contributed to writing, supervising and editing the article.
AUTHOR DISCLOSURE
No competing financial interests exist.
FUNDING INFORMATION
This work was supported by the Agència de Gestió d’Ajuts Universitaris i de Recerca, Generalitat de Catalunya (2021-SGR 00529 to AB; 2021-SGR 0488 to XN), and the Ministerio de Ciencia, Innovación y Universidades (PID2022-140354OB-I00 to XN; PID2023-148834OB-I00 to AB).