
Abstract
The first marketed gene therapy medicinal products based on adeno-associated virus (AAV-GTMP) show promise for the treatment of various diseases, including rare diseases with unmet medical needs. AAV is traditionally considered nonpathogenic to humans, is incapable of self-replication, and, after introduction into various cell types, remains primarily episomal. Several reports have examined the risks of AAV-GTMP, including the risks associated with unintended integration events of elements from the recombinant (r) AAV vector into the host genome. Such events can be one of the steps in the multistep process of tumor formation. To date, rAAV-gene therapy (GT) vectors have not been shown to induce tumors in humans or non-rodent species, and the potential for rAAV-mediated carcinogenicity in humans is still considered theoretical. Nevertheless, a critical review of publicly available scientific data on rAAV-related integration events and a contextualization of the numbers of AAV-GT vector DNA integrations with the absolute burdens of environmental, lifestyle and background tumorigenic genotoxicities is warranted. From a regulatory perspective, it is advisable to implement a long-term safety follow-up for patients who have undergone treatment with high doses of AAV-GT, in accordance with the risk-based approach that has been established for advanced therapy medicinal products.
INTRODUCTION
Gene therapy medicinal products based on adeno-associated virus (AAV-GTMP) offer great potential for the treatment of various diseases including rare diseases with limited therapeutic options, and the first of these have been centrally authorized in the European Union/European Economic Area (Table 1). In this review, scientific regulatory experts from European national competent authorities and in their role as experts of the Committee for Advanced Therapies (CAT) at the European Medicines Agency (EMA) highlight the currently available scientific data on recombinant (r) AAV (rAAV)-related integration events to support regulatory decision-making, including safety monitoring of patients treated with AAV-GTMPs. Vector integration is associated with an inherent risk of tumorigenesis, as integration can disrupt chromatin or gene structure, thereby altering gene transcription, regulation, and/or coding sequences. 1 This could result in oncogene (trans)-activation or tumor suppressor gene inactivation leading to cellular transformation. The acquisition of the malignant phenotype is thought to be a multistep process. 2 This is supported by observations that integrations close to proto-oncogenes do not necessarily lead to malignant clonal cell expansion and that malignant transformation (the process whereby a normal, metaplastic, or benign neoplastic tissue/cell becomes a cancerous tissue/cell) can occur years after the integration event. 3 Fail-safe mechanisms, intrinsic to the host cell, efficiently prevent the insurgence of cell clones harboring integrations and thus reduce the risk of insertion-mediated oncogenesis (also known as tumorigenicity or tumorigenesis). 4
Overview of Centrally Authorized AAV-GTMPs in the EU/EEA
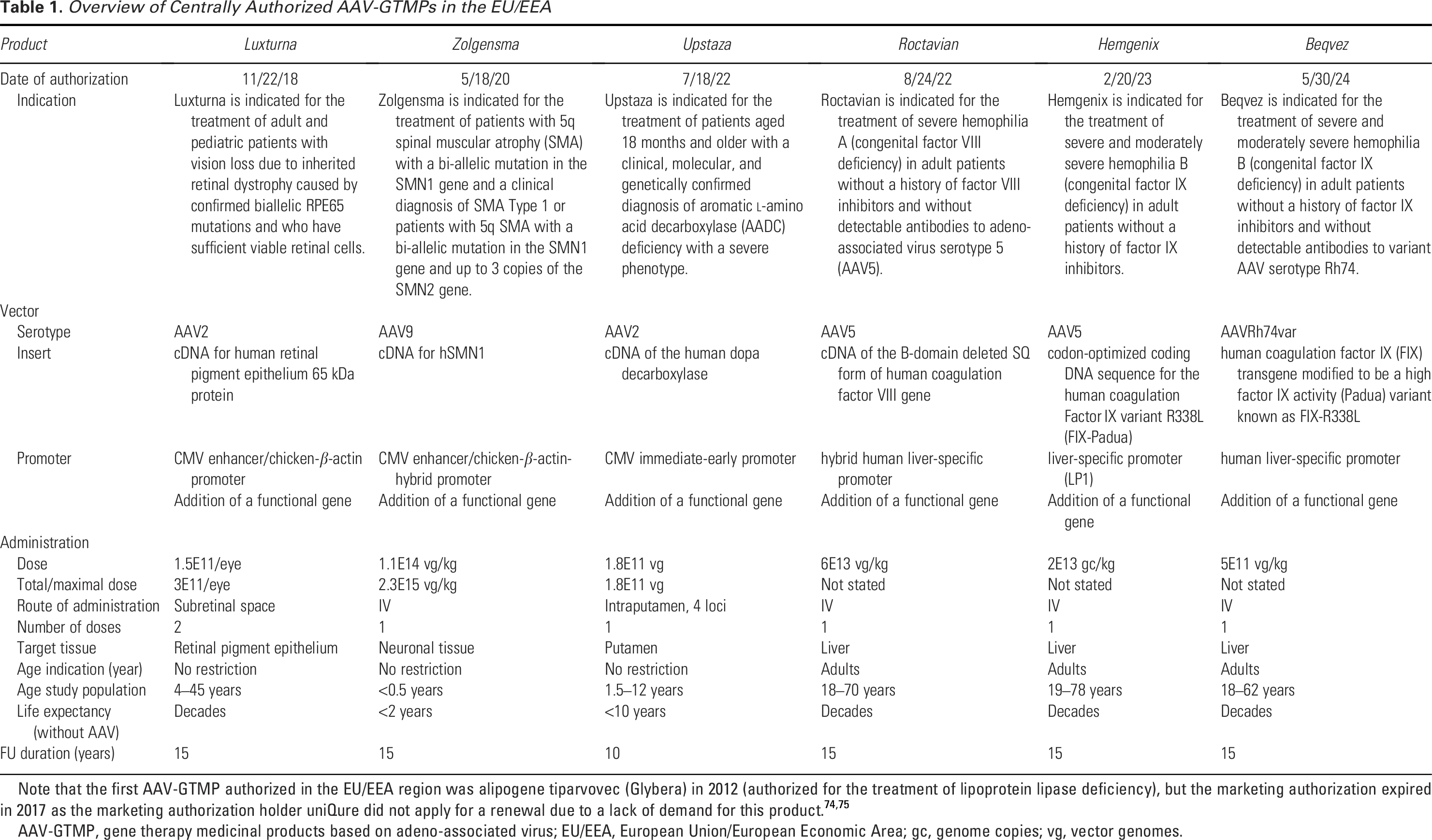
Note that the first AAV-GTMP authorized in the EU/EEA region was alipogene tiparvovec (Glybera) in 2012 (authorized for the treatment of lipoprotein lipase deficiency), but the marketing authorization expired in 2017 as the marketing authorization holder uniQure did not apply for a renewal due to a lack of demand for this product.74,75
AAV-GTMP, gene therapy medicinal products based on adeno-associated virus; EU/EEA, European Union/European Economic Area; gc, genome copies; vg, vector genomes.
Integration of exogenous DNA into a host cell’s genome has been widely described for a variety of (wild type) viruses and gene therapy (GT) vectors. In rare cases, naturally occurring viral integration has been linked to cancer (e.g., hepatitis B virus [HBV] with hepatocellular carcinoma [HCC], human papillomavirus with cervical carcinoma). For certain integrating GT vectors, specifically gamma-retroviral vectors (γ-RV), cases of tumor formation related to integration events have also been reported. 5
INSIGHTS FROM CLINICAL EXPERIENCE WITH AAV-GT VECTORS
With regard to the clinical experience with AAV-GT vectors, to date there are five reports of cancer in participants of an AAV-GT trial, mainly in the setting of hemophilia, with one of the cases reporting tumor formation in the target organ, that is, the liver. This case concerns a clinical trial subject in the HOPE-B phase 3 trial who received etranacogene dezaparvovec (Hemgenix) for the treatment of hemophilia B and developed HCC. 6 UniQure reported in a press release that vector integration in the patient’s tissue sample was extremely rare (0.027%) with integrations distributed across the genome; in addition, it was noted that integration frequencies were similar in the malignant/nonmalignant liver biopsies from this patient, and no evidence of clonal expansion or any dominant integration event was found. 7 Furthermore, these studies revealed (1) the presence of genetic mutations in tumor cells known to be associated with HCC and cancer in general, (2) the surrounding tissue was in a precancerous state, and (3) the patient had HCC-associated risk factors (infection with HCV and HBV, nonalcoholic fatty liver, advanced age, history of smoking and cancer in the family). These findings were interpreted as predisposing factors for the development of HCC, and it was considered unlikely that insertional mutagenesis of the AAV-GT vector had played a major role in the development of liver cancer in this patient. Furthermore, Symington et al. recently published safety data from a phase 1/2 hemophilia study, which included the diagnosis and detailed investigation of a serious adverse event in form of acinar cell carcinoma of the parotid gland that occurred in one 6 × 10E13 vg/kg bodyweight (bw) cohort participant during year 6 following treatment with valoctocogene roxaparvovec (Roctavian). While the detection of vector integration sites by target enrichment sequencing and the data analyses employed were appropriate for biopsy samples with a relatively low tumor content, it would appear that genomic analyses of resected parotid tissue did not reveal a causal relationship with Roctavian vector integrations. 8
The two other cases concern a report of parotid-gland carcinoma and of B-cell acute lymphoblastic leukemia; both were not considered related to treatment as no increased vector copy numbers were identified in tumor cells.9,10 In addition, Retson et al. published a case of a 16-month-old male with spinal muscular atrophy who was diagnosed with an epithelioid neoplasm of the spinal cord approximately 14 months after receiving onasemnogene abeparvovec (Zolgensma). In situ hybridization analysis detected an AAV-GT-specific nucleic acid signal broadly distributed in many but not all tumor cells. Integration site analysis on patient tumor samples failed to detect high-confidence integration sites of the respective AAV9-GTMP. 11
Two recent publications provide an overview of AAV-GT in clinical trials over the last 20 years12,13 but do not include data on the number of total treated subjects. An estimation of the number of subjects actually treated with rAAV is difficult to make as many of these studies are still ongoing, and often only planned/estimated patient enrolment is reported. Based on a targeted search in clinicaltrials.gov, using the information provided from these reviews, and some conservative assumptions, it is estimated that at least 1,500 patients have been treated to date with rAAVs (all types) within clinical studies and that approximately half of these have been treated at least 5 years ago, with approximately 100 subjects treated over 10 years ago. In addition to administration during clinical studies, patients are also treated in clinical practice with commercially available approved AAV-GTMP. Because approvals were relatively recent, the duration of follow-up of these subjects is limited, but the number of patients treated in post-marketing setting is substantial (a press release by Novartis in March 2024 stated that 3,700 patients were treated globally with Zolgensma). 14 In summary, although the clinical experience with AAV-GTMP is still limited, the number of patients is increasing as is the duration of follow-up with some patients having been treated as long as 20 years ago,12,13 and so far, no cases of AAV-GT-related insertion-mediated tumorigenesis have been reported.
INSIGHTS FROM PRECLINICAL ANIMAL STUDIES AND WILD-TYPE AAVS
As AAV-GT vectors are derived from wild-type AAVs (AAVwt) and various AAV strains exhibit liver tropism, it may be of interest whether AAVwt integration events are associated with HCC. In a small percentage of HCC cases, AAVwt integrations were noted to be located near proto-oncogenes, which could be associated with elevated transcription of proto-oncogenes in some of these tumors.15–18 Additionally, an analysis of human HCCs provided evidence of a liver-specific enhancer–promoter function in the 3′ untranslated region of the AAV2wt genome, potentially dysregulating host HCC driver genes and hence contributing to the multifactorial pathogenesis of human HCC. 19 While these studies could suggest that AAVwt integration might have contributed to the malignant transformation of liver cells, it is still premature to conclude that AAVwt has a causal role in HCC. 20 Notably, in humans, the prevalence of HCC (10 per 100,000) is much lower than the occurrence of AAV2 infection (at least 40% of the population is seropositive for AAV2wt). This is interpreted as providing arguments against a strong correlation between AAV2 infection (and potential integration) and tumor formation. 21
With regard to animal data, there are some mouse models in which HCC caused by AAV-GT-mediated transactivation of proto-oncogenes has been observed.22–27 The most prominent example of AAV-GT-related HCC is the neonatal model in which HCC was found in >70% of mice following a single intrahepatic injection of a rAAV vector. 22 The tumors in these mice were mostly caused by vector integrations mapped to Mir341. Mir341 is a 96-base pair region (coding a microRNA) contained within the Rian locus, which is highly expressed early in the neonatal period but has a low expression in juvenile/adult mice and is not present in the human genome. Integration events have also been observed outside of the Rian locus and near proto-oncogenes.3,28–30 Currently, the impact of integration events at these sites remains unclear. Yet, these data indicate that AAV-GT vectors have a certain insertional tumorigenic potential in mice.
A study in nine juvenile dogs with hemophilia A reported hepatic clonal expansion 7–10 years following intravenous (IV) administration of rAAV8 or rAAV9 vectors expressing canine FVIII. 3 In the analyzed clones, integration events were detected in genes associated with transformation in humans; however, whether the clonal expansion was caused by the integration events is not known. Importantly, no signs of nodule formation or tumorgenicity were observed in these animals, suggesting that the clonal cells still responded to environmental signals and supporting the notion that malignant transformation is a multistep process. Of note, benign, age-related hepatic nodular hyperplasia is a known phenomenon in dogs. 31 Additionally, in hemophilia B dogs, long-term follow-up after treatment with a variety of rAAV serotypes did not show signs of neoplasia.32–34 To investigate the mechanism of transduction that affects AAV-GTMP performance, a recent study in nonhuman primates (NHPs) following IV administration of AAV8 and AAVrh10 vectors with nonimmunogenic transgenes more than 2 years earlier suggested that AAV-mediated transgene expression in primate hepatocytes occurs essentially in two phases: a high but short-lived expression from episomal genomes, followed by much lower but stable expression, likely from integrated vectors. 35
HOW DOES INTEGRATION OF AAV-GTS OCCUR?
Following transduction of cells and tissues with AAV-GT vectors, the rAAV vector genome persists mostly in form of circular nonintegrated DNA concatemers. 36 However, some level of integration of the rAAV vectors into genomic DNA was found to occur in animal models as well as in humans.3,9,29,37–40 The integration sites for AAV-GT vectors are distributed throughout the genome with preferential integration into regions of open chromatin structures and near actively transcribed genes.9,41,42 Integration of AAV-GT vectors often involves concatemerization, partial deletion, or rearrangement of the vector genome.
The estimations of the integration rate for AAV-GT vectors vary depending on the study source. It can range from 0.00001% to 10% of the administered vector genomes36,43,44 across various animal models (NHP, mice, and rabbits) as well as analyzed tissues and integration site (IS) detection methods. Apart from the relative integration rate (the proportion of the administered vector genomes that integrate into the host genome), the absolute integration burden per transduced target cell or tissue/organ can be calculated by applying the vector copy number (VCN) and the integration rate per cell as well as the total tissue or organ cellularity, respectively. This way, absolute integration numbers were, for example, calculated with relative integration rates available for the approved AAV-GT Roctavian and Hemgenix in the supplementary information to this review (and these absolute integration burdens were further contextualized with absolute burdens of common environmental, lifestyle, and background tumorigenic genotoxicities). For example, relative rAAV genome integration frequencies in host liver cells of cynomolgus monkeys and patients after Roctavian or Hemgenix administration were reported to range between 1.55E-03 and <3.00E-04 integrations per liver cell and between 2.10E-05 (Hemgenix, at a VCN of 11 per liver cell) and 6.10E-05 (Roctavian, at a VCN of 24 per liver cell) integrations per liver cell transducing vector genome.9,40 These values can be used to estimate absolute rAAV vector DNA integration burdens in the liver of patients (as further elaborated in the Supplementary Data), which would range from 9.71E07 to 6.05E08.
AAVwt is also known to integrate into genomic DNA. However, the integration pattern and the associated tumorigenic potential have only limited relevance to evaluating the risks of AAV-GT vectors because of differences between AAV-GT vectors and AAVwt. Mapping studies in cultured human cells showed that AAV2wt tends to integrate in open chromatin regions near consensus Rep-binding elements.41,45 However, AAV-GT vectors lack Rep and, as a consequence, integrate into genomic DNA far less efficiently than AAVwt, although the viral load of AAV-GT is different and presumably much higher than during an infection with AAVwt. Furthermore, the components regulating gene expression (promotor/enhancers) in the AAV-GT are also manipulated thus potentially impacting the outcome of an integration event on the expression of nearby genes.
In neonatal mice, rAAV integrations target not only transcriptionally active genes such as Alb and Afp but also the Rian locus in which integrations of rAAV are associated with murine 22 HCCs. In murine HCCs, rAAV is specifically present in the Mir341 site of the Rian locus that is absent from all vertebrate genomes except for those of mice and rats. 46 It has been proposed that this site has an unusual secondary structure during or after unwinding due to the number of copies of quadra-nucleotide repeats (CCGT and GGCT) and CpG motifs. 46 This specific structure could serve as a preferred substrate for rAAV recombination.
Of note also for other GT vectors (e.g., lentiviral vector [LVV], γ-RV), semi-random integration into the host genome with a preference for active regions has been shown. However, differences in the integration profile have been reported based on the used GT vector type, which is most likely related to the difference in cellular interaction partners that are involved in the integration process. For example, γ-RV preferentially interacts with histones, while LVV generally interacts with transcription machinery.47,48 So while some general insights and concepts of the risks associated with genomic integration can be learned from other (integrative) GT vectors, such as LVV and γ-RV, one should be cautious with over-interpretations. Each vector (type) should be evaluated based on its own integration characteristics.
WHAT MIGHT INFLUENCE THE RISK OF INSERTION-MEDIATED TUMORIGENICITY BY AAV-GT VECTORS?
Theoretically, any integration event can predispose a cell to transformation. However, the risk of tumor formation is expected to be related to the number of integration events, which is assumed to be directly related to the dose of the vector. Vector dose is generally expressed as viral genome copies, and dose levels for AAV-GTs vary widely and depend on route (or location) of delivery. Systemically administered AAV-GTs have been studied at doses ranging from 3.5E13 to 1.5E17 vector genome copies (total dose per patient), while for targeted/local delivery, dose levels of 5.8E9 to 7.5E15 have been used. 13
As integration generally occurs in transcription-active regions and at chromosomal breaks, the transcription activity and the level of chromosomal breaks can be anticipated to affect the frequency of integration and the insertion profile. For example, the incidence of HCC development following rAAV vector administration in mice was reported to be highest in neonatal mice.49–51 This was attributed to the fact that the Rian locus is most transcriptionally active during early stages of the liver development. 52 Another example was reported in a study by Dalwadi et al., which suggests that a preexisting pathology (induced liver damage) may impact the frequency of rAAV vector-induced HCC with incidences in HCC formation as high as 50% after administering the rAAV vector to mice that received a high-fat diet. 29 The authors highlighted that increased hepatocyte proliferation, coupled with inflammation, contributed to a higher incidence of HCC. It can thus be argued that under stress conditions inducing DNA breaks, the frequency of integration events may increase. Notably, exposure to genome-targeted nucleases that produce double-strand breaks is seen to lead to higher levels of AAV-GT vector integration or generates frequent concatemeric insertions of viral vectors at the target site.53–55 The combination of increased hepatocyte proliferation rates and increased frequency of integration events could potentially work synergistically to favor tumor formation. This synergism is known for chemical tumorigenesis and may be applicable to insertion-mediated tumorigenesis. It is important to note that while administration of an AAV-GT vector itself is often accompanied by some degree of liver inflammation, the level and duration of inflammation differ between patients and may also be influenced by the AAV-GT vector dose administered, potentially existing liver condition (e.g., nonfatty liver disease) and the use of (concomitant or triggered) anti-inflammatory therapy.
The serotype of the rAAV vector influences vector tropism and in vivo biodistribution and thus has an impact on tissue-specific disposition of the AAV-GT vector and subsequent transduction efficiency. In addition, rAAV capsid proteins can be engineered to display a more selective or desired tissue tropism. While this may not affect the intrinsic properties of the rAAV to integrate, the ability to integrate may be changed due to (differential) properties of the target cell/tissue. As a result, the vector serotype and/or biodistribution pattern may impact the risk of AAV-GT vector-integration and integration-mediated tumorigenicity. Also for other GT vectors, such as LVV, it has been shown that the integration profile varies in different target cells, reflecting an impact of cell properties on vector integration. 56
Studies in mice demonstrate that HCC can be induced by rAAV vector-mediated transactivation of proto-oncogenes.22,23,25,28 Indeed, in a recent and yet unpublished study on the genomic architecture of carcinogenic rAAV integrations in murine HCCs (with all rAAV integrations mapped to the Rian locus), all rAAV integrations included AAV enhancers and/or transgene promotors, suggesting that the rAAV transgene enhancer drives proximal transcript upregulation and carcinogenesis after rAAV integration. 57 Notably, in the same study only a minority (3 out of 24) of the integrations contained transgene coding sequences, suggesting that targeting the rAAV transgene to prove or disprove a genotoxic event in a tumor is not a sufficient approach. Additionally, the type of enhancer-promotor used is anticipated to affect the impact of integration on (trans-) activation of nearby genes and thus on integration-mediated tumor formation. This is exemplified by yet another recent and unpublished investigation, which reports that the enhancer promoter used in Zolgensma induced HCCs in mice but not the endogenous native hSMN1 promoter, supporting that regulatory elements in rAAV-GT can clearly vary in their genotoxic potential. 58 As a certain level of (micro)homology with host genomic sequences could facilitate integration at chromosomal breaks, the nucleotide sequences used in the vector may also impact the integration profile and possibly also the frequency of integration.
Until now, development of AAV-GT has largely been focusing on diseases where liver is the relevant target organ. As a consequence, the most extensive knowledge on AAV-GT-induced toxicity, including insertional mutagenesis (i.e., integration risk), has been obtained with rAAV serotypes preferentially targeting liver tissue. Whether these studies may be useful to identify general risk factors for AAV-GT irrespective of tissue distribution or only for those AAV-GT vectors targeted to the liver is not yet known; however, emerging data on integration using other tissue targeting serotypes may provide valuable insights. In addition to the AAV-GT vector itself, its manufacture may need to be considered for the risk of integration events. The method of vector production may affect the number and type of viral genome rearrangements present in the product, and with that the probability of insertion. Furthermore, as chimeric AAV/plasmid sequences have been found integrated in genomic DNA,59,60 the presence of DNA impurities from the plasmids or production cell may need to be considered as well. Indeed, a few percent of AAV-GT vector particles were found to contain human DNA fragments acquired randomly from the host/production cells used to manufacture the vector.61,62 In particular, as the production cell is likely to be an immortalized cell, (viral) oncogenes from these immortalized cell lines could end up as DNA impurity in the AAV-GT vector. Currently, there is insufficient information to evaluate the tumorigenic risks from the presence of viral oncogenes in the production cells during the manufacturing of rAAV vectors, and this risk factor should be considered in the choice of production cell lines.
AAV-GT vectors are increasingly used for genome editing, either carrying genome editing components or to serve as DNA donors for homologous recombination. 63 When used together, genome-targeted nucleases and AAV vector donor templates synergize to produce relatively high levels of genome editing. 64 As noted above, such combination with nucleases could also lead to increased levels of unwanted AAV-GT vector integration.53,54,65 Other risk factors include the potential for sex-specific differences in AAV-induced tumorigenicity. Female mice were less susceptible than male mice to AAV-induced HCC, while male mice with nonalcoholic fatty liver disease treated with estrogen displayed less inflammation and immune exhaustion associated with tumorigenicity compared with those not receiving estrogen. 29
In conclusion, while the relationship between dose and the risk of integration can be expected, other risk factors are also likely to affect the integration profile and/or frequency of integration and may contribute to this risk. These include the type and (diseased) state of the tissue, the design of the vector (affecting distribution and tissue tropism, choice of promotor/enhancer, presence of [micro] homologies), and vector manufacturing processes and outcomes. Therefore, the extrapolation of results from one AAV-GT vector to another should be done with caution. Additionally, tissue tropism of AAV-GT vectors can vary across species, complicating interspecies comparisons and extrapolations of animal data to humans. 66
CAN THE CLINICAL RISK OF INSERTION-MEDIATED TUMORIGENESIS FOR AN AAV-GTMP BE ESTIMATED BASED ON NONCLINICAL AND CLINICAL SAFETY STUDIES?
Integration of AAV-GT vectors and thus their integration profile can be observed in vitro, both qualitatively and quantitatively. These assays contribute to the understanding of the integration events of the vector tested, but the use of the data to extrapolate and estimate the clinical risk of insertion-mediated tumor formation is hampered. This is not only because quantitative information is lost in translation, but also because integration is only a first step in the process of tumor formation and the next steps in the transformation process cannot be studied in vitro so far. Also, the impact of tissue distribution of the GT vector or disease pathology is difficult to evaluate with these assays.
In vivo studies/animal models to study tumorigenicity/oncogenicity of AAV-GT vectors have the advantage that biodistribution of the AAV-GT is an integral part of the study and that multiple tissues can be evaluated for integrative and oncogenic events. As such, potential differences in integration patterns between tissues and in time can be studied, although it should be noted that the tropism and hence biodistribution may differ between species. Furthermore, risk factors contributing to the risk of tumorigenesis may be identified. In this context the mouse model with preferential integration in the Rian locus early in development, as discussed above, is noted.
However, the sensitivity of animal models to detect oncogenic events can be questioned, especially when integration events are infrequent and lack preferential integration sites. To detect oncogenic events, large sample sizes with assessments after a long follow-up (FU) would be needed. Given the long-term persistence of rAAV vectors and reports of hepatocyte clonal expansion 3 or HCC development later in life in dogs and mice, respectively, lifelong monitoring of animals following dosing may be informative. However, given the difficulties with extrapolation into a clinical risk, a general recommendation for lifelong FU in animals is not foreseen. There are some reports of long-term FU of nonrodent animal studies, even up to 6–10 years,3,25,32,43,67–69 but animal numbers are often limited, and given the lifespans of these animals, it is currently unclear whether these animals were evaluated long enough to determine the incidence of rAAV vector-induced tumorigenic events. It took years to develop and detect clonal dominance in the hemophilic dogs, 3 and it remains uncertain whether integration was the cause behind clonal expansion as this has not yet been associated with oncogenic changes. A model more prone to tumorigenesis could be used to increase the frequency of tumor formation, but it is not clear what type of model could be used and how this could be translated to a clinically relevant risk assessment. A recent study in a human liver chimeric, transgene-free Il2rg-/−/Rag2−/−/Fah−/−/Aavr−/− (TIRFA) mouse model combines liver humanization with ablation of the AAV receptor, rendering murine cells insensitive to AAV transduction. Using this chimeric TIRFA mouse model, Barzi et al. showed higher transduction of AAV-8 and AAV-9 serotypes in primary human hepatocytes compared with humanized mice with wild-type AAV-receptor. Such mouse models may allow prediction of AAV gene transfer efficiency and the study of AAV vector biology in a nonclinical setting, including tumorigenic events. 70
Altogether, both in vitro and in vivo nonclinical studies have their limitations in predicting the tumorigenic potential of AAV-based GTs and uncertainties persist in the interpretation and the relevance of carcinogenic signals observed in non-clinical studies.
Clinical information on AAV-GT vector insertion and potential integration-mediated tumor formation would be most informative, but collection of such data in the clinical setting is accompanied by both ethical and technical challenges. An important aspect is tissue accessibility, and (frequent) biopsies of the target tissue (often liver) are generally not possible. In principle, although integration site analysis on more easily accessible biological samples (e.g., plasma or other fluids) might be an option, it is not yet clear how this would relate to the target tissue and to any transformation. Recently, in a clinical trial based on integrative vectors, the usefulness of plasma derived cell-free DNA (cfDNA) as a potential biomarker for early identification of expanding clones growing in solid organs was studied. 71 Preliminary data indicated the feasibility of the approach in identifying rAAV integration in cfDNA purified from blood plasma of rAAV treated NHPs, but further research is required to validate this assay for its clinical use.27,71
Other assessments for tumorigenicity could be performed at regular intervals for early identification of any tumor formation (ultrasound of target tissue or positron emission tomography/computed tomography scans). This does pose a substantial burden to the patient (both logistically and the latter in terms of radiation dose) and the health care system. Since the risk of insertion-mediated tumorigenesis is currently considered to be low, regular evaluation for tumorigenicity is not justifiable in clinical practice. Imaging methods could be included in clinical trials, but as the number of patients in AAV-GT studies is often limited, the sensitivity to detect tumor formation would be limited as well, in particular for patients with longer term follow-up. However, since the cfDNA analysis (next-generation sequencing liquid biopsy) is becoming more used in routine clinical practice, this can be proposed to be validated for follow-up of AAV-GT-treated patients, favorably coupled with germ-line analysis of DNA repair genes. Given the many unknowns, it needs to be determined whether pooling data across patient populations, doses, vectors, and routes of administration is possible.
CONCLUSION FOR PATIENT SAFETY FOLLOW-UP AFTER ADMINISTRATION OF AAV-GTMPS USING THE RISK-BASED APPROACH FOR ADVANCED THERAPY MEDICINAL PRODUCTS
Based on the above considerations, a “zero risk” for insertional tumorigenicity cannot be assumed. Their absolute magnitude can—to some extent—provide an estimate of potency when contextualized to absolute numbers of well-established tumorigenic genotoxicities. To this end, the absolute numbers of AAV-GT vector DNA integrations were contextualized with absolute burdens of common environmental, lifestyle, and background tumorigenic genotoxicities. As illustrated in Supplementary Data, this contextualization indicates that the absolute numbers of AAV-GT vector DNA integrations, such as expected in the liver of patients after Roctavian or Hemgenix administration, might be considerably lower (up to five to six orders of magnitude) than excessive mutations found in organs that are or have been chronically exposed to genotoxic carcinogens. Even if the risk is low, it is reasonable to assume that there may be a potential risk to patients treated with AAV-GTs. Given that an integration event may be one step in a multistep process leading to tumor formation after an unknown latency period, it is necessary to have an adequate patient follow-up from a regulatory perspective.
The duration of clinical FU for safety should be dependent on identified risk factors and the anticipated time until occurrence of (delayed) adverse events (latency) 72 rather than requiring a general lifelong follow-up. Given the uncertainties associated with the long-term effect of AAV-GT, this follow-up should be adequate to collect both safety and efficacy data but should be proportionate to the anticipated risks/effects. It is currently not possible to estimate the risk of tumor formation on the basis of manufacturing and nonclinical data. The question of which considerations could assist in defining a well-balanced, flexible, and, at the same time, data-driven approach in the benefit–risk evaluations of AAV-GTMPs remains challenging to answer.
As tumor formation can take years, a 15-year follow-up is recommended for patients treated with high doses of AAV-GT vectors, such as administered, for example, Roctavian (6E13 vg/kg bw), Hemgenix (2E13 vg/kg bw), and Zolgensma (1E14 vg/kg bw). Deviation from this general rule could be justified by a developer on a case-by-case basis using the flexible regulatory tool of the risk-based approach for advanced therapy medicinal products. Thus, product-specific quality, nonclinical, and clinical risk factors should be considered, such as dose level, delivery route (targeted or systemic), the vector design (type of enhancer-promoter), whether it encodes genome editing components, the proliferative activity of the (target) tissue, the need for concomitant (mutagenic) therapy during the viremia phase, and the patient’s age range, comorbidities, and life expectancy. 73 In any case, a minimum safety FU period of at least 5 years is expected for patients treated with an AAV-GT. 72 Finally, and most importantly, patients should be well informed about the potential tumorigenic risk of AAV-GT products before treatment is initiated.
AUTHORS’ CONTRIBUTIONS
Conceptualization: E.F. and M.W. Investigation: E.F. and M.W. Writing—original draft: E.F., M.W., and M.S.-L. Writing—review and editing: E.F., M.W., M.S.-L., V.C.-C., P.C., and I.R.
Footnotes
ACKNOWLEDGMENTS
The authors thank Carla Herberts, Jessica Hartmann, and Frederic Thalheimer for their support of this project and instrumental contributions. They also thank Renate König, Zsófia Nacsa, and Arezoo Jamali for their assistance with article preparation and graphics.
FUNDING INFORMATION
This study has not received any funding.
PEER REVIEW INFORMATION
The authors thank the EMA for their contribution to the internal peer review of this work.
DISCLAIMER
The views expressed in this article are the personal views of the authors and may not be understood or quoted as being made on behalf of or reflecting the position of the EMA or one of its committees or working parties or the National Agencies of the individual European member states.
AUTHOR DISCLOSURE
All authors have no competing interests.
Supplemental Material
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.