
Abstract
Manganese is a ubiquitous, essential trace element and a common ingredient of joint supplement tablets. Little information is known about the inherent toxic potential if ingested at higher doses. A 5-year-old female spayed Pug dog presented for evaluation of vomiting and ataxia after accidental ingestion of approximately 100 joint supplement tablets. The dog developed acute hepatic failure and was euthanized 6 days after presentation due to progression of the disease. Necropsy showed severe acute hepatic necrosis. Liver and kidney samples were submitted for toxicology analysis, results of which showed severely elevated manganese concentrations in the liver and kidneys.
Keywords
Manganese (Mn) is both a ubiquitous essential trace element and a toxic metal if ingested at higher doses. Food is the major source of Mn in human beings and animals. Usually, homeostatic mechanisms limit the intestinal absorption to only 1–5% of the normal dietary intake with less than 1% retained after biliary and intestinal excretion while urinary excretion is fairly low. 2
Manganese is required for amino acid, lipid, protein, and carbohydrate metabolism. It also serves as an important cofactor for many enzymes including hexokinase, superoxide dismutase, xanthine oxidase, and pyruvate carboxylase, many of which are localized in mitochondria.2,23 Manganese is evenly distributed throughout the body. Increased Mn concentrations are mostly found in tissues with both high energy demand and mitochondria such as brain, liver, kidney, pancreas, and bone as well as in highly pigmented tissue. 23
Manganese toxicity has been reported in human beings and in experimental animal models. 28 Chronic Mn intoxication (manganism) due to prolonged inhalation of Mn dust or fumes in miners, welders, or battery manufacturers historically causes neurologic disease in the form of a Parkinson’s-like bradykinetic-hypertonic syndrome.2,4,18 Long-term parenteral nutrition 9 and chronic Mn exposure through contaminated water have also resulted in neurologic disease in human beings. 18 Acute Mn intoxication is rare and has been scarcely reported in the literature. Other human case reports include a patient that developed severe pancreatitis after hemodialysis due to a dialysate contaminated with Mn, 24 acute Mn pneumonitis after inhalation of Mn-containing dust or fumes, 11 and acute fatal Mn intoxication after accidental, massive oral ingestion. 22 Experimental, intravenous Mn administration in Beagle dogs resulted in severe hepatotoxicosis and hypotension. 15 Acute enteral Mn intoxication has not been reported in animals.
A 5-year-old female spayed Pug dog, weighing 5.8 kg, presented to the University of California–Davis Veterinary Medical Teaching Hospital (Davis, California) emergency service for evaluation of vomiting and ataxia after accidental ingestion of approximately 100 joint supplement tablets. Approximately 6–12 hr prior to presentation, the dog ingested approximately 60 g of glucosamine hydrochloride (10.3 g/kg), 25 g of methylsulfonylmethane (4.3 g/kg), 30 g of sodium chondroitin sulfate (5.2 g/kg), and 500 mg of manganese ascorbate (86 mg/kg).
On physical examination, the dog was obtunded with pelvic limb ataxia, in mild hypovolemic shock, and was approximately 5% dehydrated. Other physical exam parameters included mild tachycardia and hypothermia, and the dog continued to vomit multiple times during the physical exam. Venous blood gas and electrolyte analysis showed a mixed metabolic and respiratory acidosis with hyperchloremia (143 mEq/l), hypernatremia (166 mEq/l), and hyperlactatemia (2.9 mmol/l). Initial therapy included intravenous crystalloid fluids, and antinausea and gastroprotectant medications (ondansetron, famotidine). The following morning, 12 hr after initial presentation, the dog appeared brighter, and the ataxia and conscious proprioceptive deficits had resolved. The results of a biochemistry panel showed resolution of the hypernatremia and hyperchloremia but elevation of alkaline phosphatase (ALP), alanine aminotransferase (ALT), aspartate aminotransferase (AST), creatinine kinase (CK), gamma-glutamyl transferase (GGT), and total bilirubin, while blood urea nitrogen (BUN), creatinine, total protein, and albumin were mildly decreased (Table 1). Continued progression of hepatic failure (Table 1) was observed over the next 6 days of hospitalization prompting humane euthanasia of the dog due to the poor prognosis and financial constraints of the client.
Serial biochemistry and coagulation panel results at various time points after initial presentation.*
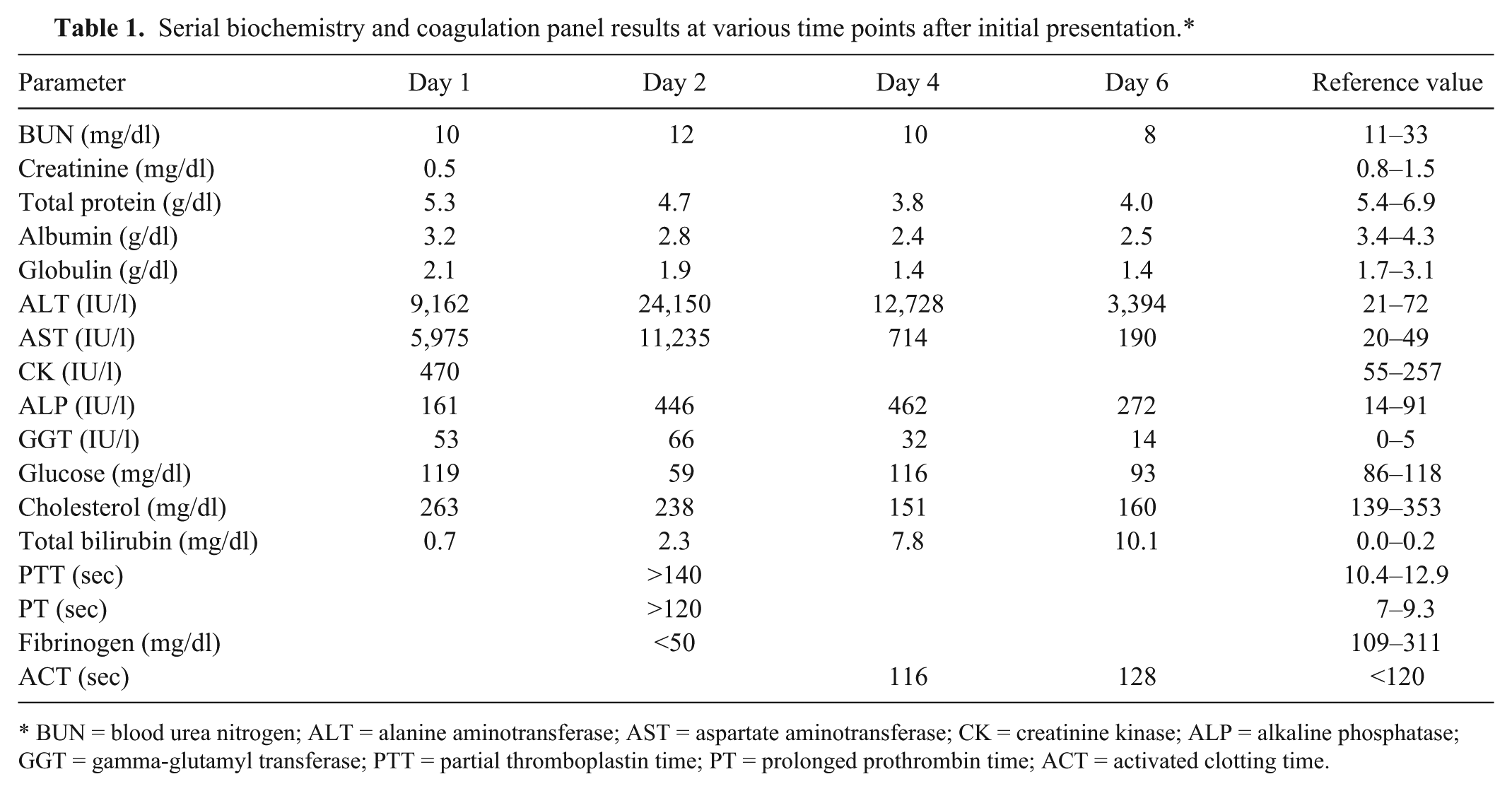
BUN = blood urea nitrogen; ALT = alanine aminotransferase; AST = aspartate aminotransferase; CK = creatinine kinase; ALP = alkaline phosphatase; GGT = gamma-glutamyl transferase; PTT = partial thromboplastin time; PT = prolonged prothrombin time; ACT = activated clotting time.
On postmortem examination, the oral and vulvar mucous membranes, sclera, and skin of the pinnae were markedly icteric. There was severe, regionally extensive hemorrhage within the subcutis of the ventral abdomen and axillary and inguinal regions, the serosal surface of the stomach, esophagus, and small intestine, and the wall of the gall bladder. The abdominal cavity contained approximately 300 ml of clear, yellow, watery fluid. The liver appeared subjectively small, weighing 149 g (2.1% of body weight; normal percentage is approximately 3.5%), and was diffusely golden yellow to tan with a subtle red, reticulated pattern and dozens of variably sized, irregularly shaped areas of red, shallowly depressed parenchyma (Fig. 1A).
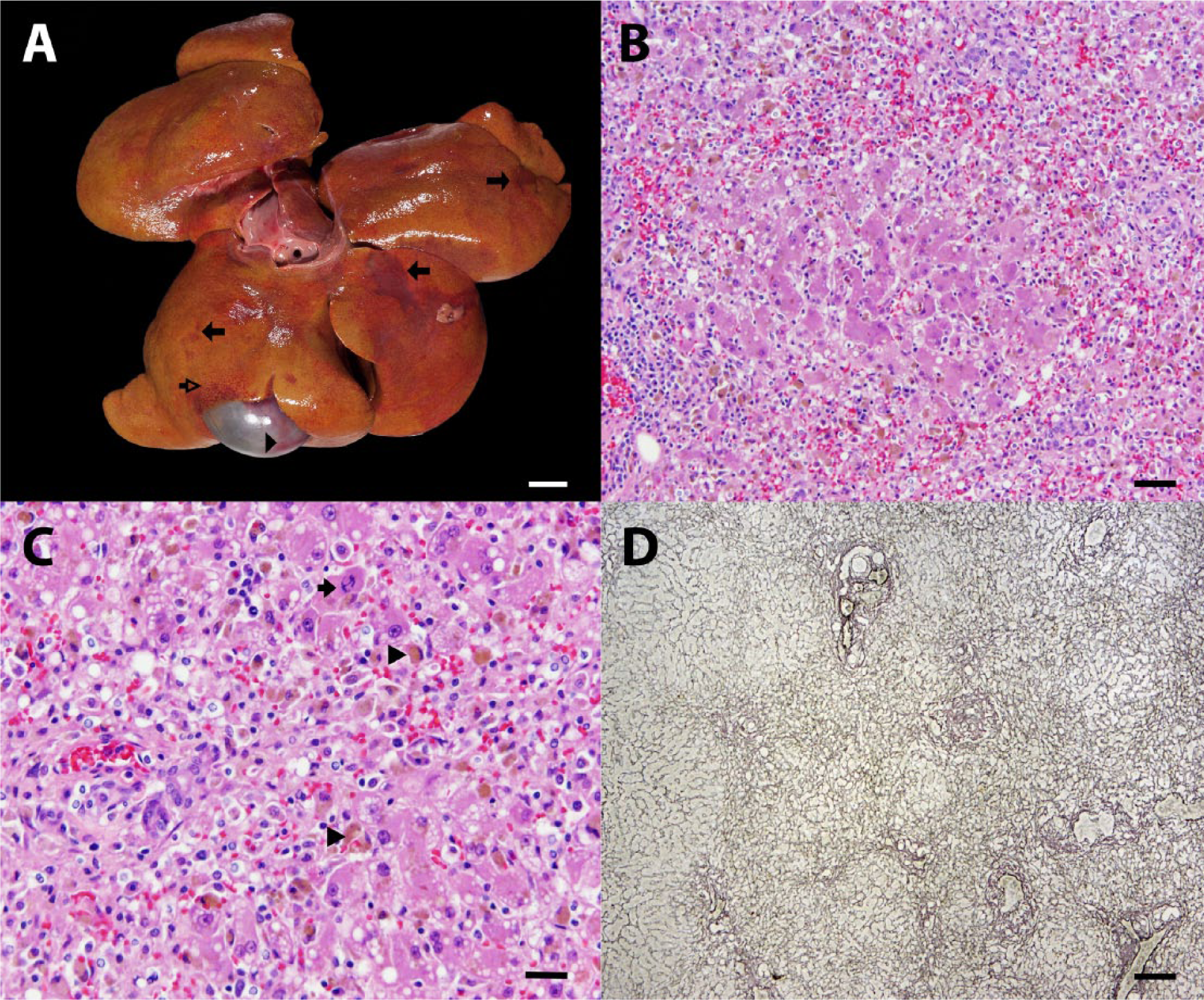
Five-year-old female spayed Pug dog.
Histological examination of the liver revealed subacute to chronic, portal-to-portal bridging, liquefactive necrosis with lobular collapse, lymphoplasmacytic hepatitis, macrovesicular lipidosis, and intracanalicular bile stasis. Hepatic necrosis was characterized by loss of hepatocytes in periportal locations and replacement of the normal hepatic chords by numerous erythrocytes, mixed mononuclear inflammatory cells, and ceroid-laden macrophages (Fig. 1B–D). Reticulin staining revealed condensation and collapse of the periportal reticulin network. Remaining hepatocytes throughout the liver contained multiple, variably sized, clear vacuoles within their cytoplasm, and frequent, dark brown, canalicular bile plugs. Ongoing hepatic necrosis was evidenced by scattered hepatocytes that were individualized and round with hypereosinophilic cytoplasm and a pyknotic or karyorrhectic nucleus. Mild hepatic regeneration was present and characterized by binucleate hepatocytes and rare mitotic figures. Throughout all portions of the liver (portal, periportal, and centrilobular), there were moderate numbers of lymphocytes and plasma cells with lesser neutrophils. Within the kidney, there was mild, acute, and subacute tubular necrosis with regeneration, and abundant glomerular and tubular proteinosis. Affected tubules were lined by hypereosinophilic epithelial cells with pyknotic nuclei that had occasionally sloughed into the tubular lumen (ongoing necrosis), flattened and attenuated epithelium (prior necrosis), or plump epithelial cells with a large nucleus and open chromatin (regenerating epithelium). The urinary space within glomeruli and proximal convoluted tubules was often filled with abundant foamy, eosinophilic material (protein). Immunohistochemical staining of both the liver and kidney with Ki-67 antibody showed a mildly increased proliferative fraction (approximately 5% of hepatocytes and 1–3 tubular epithelial cells/tubule) indicative of early hepatic and renal tubular regeneration. Other findings included capsular splenic fibrosis, nodular adrenal cortical hyperplasia, and mild chronic serositis (stomach) with intralesional plant material. Liver and kidney samples were submitted for toxicology analysis, results of which showed high Mn concentrations in the liver (60 mg/kg wet weight, reference range = 3–5 mg/kg) and kidney (10 mg/kg wet weight, reference range = 1.2–1.8 mg/kg).
The current study confirms a case of Mn poisoning in a dog subsequent to an overdose of joint supplement tablets, highlighting the importance of the toxic potential of this substance. As this essential trace element is a common ingredient of joint supplements often administered to dogs for osteoarthritis, it is important to understand the properties and the toxic potential of Mn.
Among its many vital functions, Mn is essential for normal skeletal development as it is the preferred cofactor of glycosyltransferases. These enzymes are required for synthesis of proteoglycans that are needed for the development of healthy cartilage and bone. 14 In human beings and animals, experimental Mn deficiency led to bone and growth abnormalities. 28 For this reason, Mn is a common ingredient in supplements for bone and joint health for people as well as for animals. The joint supplement tablet that was ingested in the current case report contained a combination of 600 mg of glucosamine hydrochloride, 250 mg of methylsulfonylmethane, 300 mg of sodium chondroitin sulfate, and 5 mg of Mn ascorbate.
The supplement ingested contained Mn ascorbate. Chelation with reducing organic acids such as ascorbate theoretically increase Mn absorption by reducing Mn to its lowest oxidation state (2+), which is considered the most absorbable oxidation state.2,28 Indeed, ascorbic acid significantly increased mean Mn retention in people 17 ; however, the acid had no effect on absorption of Mn when added to a cow milk formula for human beings. 8 It is possible that the combination of Mn with ascorbate in our particular joint formulation accelerated the absorption of Mn and lead to the fulminant enteral intoxication. Alternatively, this formulation in combination with increased voluntary water intake may have led to an increased absorption of the Mn in the dog described herein. The former was speculated to be the cause for the fatal enteral Mn intoxication that was reported in a human patient. 22
Nutritional Mn requirements in the dog are estimated to be 0.16 mg/kg/day with an estimated bioavailability, including absorption efficiency, of 10%. 20 This is considerably low compared to Mn requirements that range from 2.5–5.0 mg/kg in human beings, 20 mg/kg in small rodents, to as high as 600 mg/kg in cattle or poultry. 28 The dog in the current case report ingested a Mn dose of 86 mg/kg, which is 538 times the recommended dose per day. Thus when considering the relatively low nutritional Mn requirements for dogs compared to rats, which have 125 times higher Mn requirements, a median lethal dose (LD50) dose for Mn in dogs is theoretically expected to be much lower than that of rats. There is no published LD50 dose for ingested Mn in dogs. The published LD50 dose for ingested manganese acetate in rats is 3,730 mg/kg. 20
Subacute Mn toxicosis was induced in Beagle dogs that received a parental dose of 16 mg/kg/day of manganese chloride over 4 consecutive days. 15 The dose resulted in death or moribund sacrifice of all the dogs within 4 days. Blood work showed marked elevations in liver enzymes, and necropsy confirmed massive hepatocellular necrosis, periportal hemorrhages, and mild biliary epithelial hyperplasia. The authors of the Beagle study concluded that dogs are inherently sensitive to the toxic effects of Mn, especially when compared to rats, which is further supported by the fact that parenteral administration of a Mn dose to cause hepatocellular necrosis is approximately 15 times higher in rats compared to dogs. 15
Interestingly, an acute, fatal enteral Mn intoxication has been reported in a human patient that accidentally ingested a large amount of hydrated manganese sulfate due to a pharmaceutical preparation error. 22 Autopsy of this patient found very high tissue levels of Mn. In fact, the liver and kidneys contained 63.4 and 25.5 mg/kg of Mn, respectively; such concentrations in the liver were similar to what was found in the canine patient described herein. 22 Histologic changes from the Pug are consistent with what was found in this human overdose case.
The pathologic findings of portal-to-portal bridging hepatic necrosis and renal tubular epithelial cell necrosis in the current case are consistent with exposure to a direct-acting toxin.19,26 Furthermore, these lesions as well as the serum biochemistry and coagulation alterations (elevated ALT, AST, ALP, and total bilirubin, and prolonged prothrombin time and partial thromboplastin time; Table 1) are also remarkably similar to those described in previously reported cases of Mn toxicosis in human beings and dogs.15,22 The subacute to chronic timeframe of the lesions, as evidenced by mononuclear cell infiltration, ceroid accumulation in macrophages, and early hepatocyte and renal epithelial cell regeneration, also supports the clinical history of suspected toxin ingestion 7 days prior to microscopic examination of the tissues. Additionally, the diagnosis of Mn intoxication is supported by toxicological analysis of the liver and kidneys, which revealed tissue levels of Mn that were 12 and 5.5 times higher than the adequate levels for these organs, respectively. Furthermore, tissue levels in the present case are almost identical with tissue levels of the previously reported human case with fatal, enteral Mn intoxication. 22
Accumulation of Mn in the liver can result in liver damage, restricting the rate of excretion of Mn and exacerbating Mn toxicity. 6 As such, it is possible that preexisting hepatic disease in the dog in the present case report may have impaired hepatic excretion and increased susceptibility to the toxic effects of Mn. However, the dog in the current report had no history of liver disease, had no histological evidence of hepatic disease other than the necrosis and appropriate inflammatory response that was attributable to toxin exposure, nor did recent, previous blood work show elevations of hepatocellular and/or hepatobiliary enzyme elevations.
Given the common use of joint supplements in dogs with osteoarthritis and the fact that the current report describes a dog with fatal Mn intoxication after ingestion of an overdose of Mn ascorbate, it is tempting to assume that Mn as a supplement is fairly safe at current recommended dosages. However, a 2010 letter to the editor from the American Society for the prevention of cruelty to Animals, Animal Poison Control Center, reported hepatic damage in 21 dogs following accidental ingestion of overdoses of various joint supplements. 16 Unfortunately, Mn was only listed as a minor ingredient with no information on exact concentration or dosage ingested. Laboratory abnormalities, including elevations of ALT and ALP, were seen as early as 5 hr after ingestion. Reported clinical signs resemble findings in the current case, calling into question whether it is the glucosamine or Mn responsible for the clinical signs observed in those 21 dogs.
While the elevated Mn levels in the dog described herein are supportive of Mn as the cause of disease, association does not demonstrate causation. It is unknown if one of the other ingested components such as the glucosamine hydrochloride, methylsulfonylmethane, or sodium chondroitin sulfate had toxic effects that caused the observed clinical and pathological findings.
A case report in a human patient reported steroid-responsive, severe autoimmune hepatitis after intake of a supplement containing glucosamine and chondroitin, 27 while another human patient developed cholestatic hepatitis likely of allergic origin. 21 Furthermore, a 2013 case study described temporary elevations of ALT and a skin rash in 2 patients with chronic liver disease after long-term ingestion of supplements containing glucosamine and chondroitin; liver biopsies were not performed. The results should therefore be interpreted with caution given the fact that both patients were elderly with chronic hepatitis C. 7 The mechanism for proposed glucosamine-induced hepatotoxicity in human beings is unknown but is presumed to be idiosyncratic in nature (Committee on Toxicity: 2009, Statement on glucosamine and hepatotoxicity. Available at: http://cot.food.gov.uk/cotstatements/cotstatementsyrs/cotstatements2009/cot200901).
Hyperglycemia and insulin resistance and a diabetic-like state have been infrequently reported in rats after administration of glucosamine. 10 However, a 2011 published work in rats did not confirm hyperglycemia and insulin resistance but interestingly glucosamine supplementation promoted endoplasmic reticulum stress, hepatic steatosis, and accelerated atherogenesis. 3 A Cochrane review evaluating glucosamine therapy for treatment of osteoarthritis reported that glucosamine was as safe as a placebo in terms of number of participants reporting adverse effects. 25 In veterinary medicine, polyuria and polydipsia was reported in a Golden Retriever that received 1,000 mg of glucosamine per day. The polyuria and polydipsia resolved after the glucosamine dose was lowered to 500 mg/day; no laboratory work was available. It is therefore unclear if the glucosamine supplement or other systemic disease contributed to the polyuria and polydipsia. 5 In rats and mice, the acute, oral LD50 of glucosamine is >8 g/kg and 15 g/kg, respectively, which is the LD50 of a practically nontoxic substance based on the Hodge and Sterner scale.1,12 Dogs fed glucosamine at 159–2149 mg/kg showed no adverse effects. 1 Furthermore, no adverse effects have been reported for chondroitin in the literature. It therefore seems unlikely that the glucosamine and chondroitin caused observed fulminant hepatic failure in the current case. Methylsulfonylmethane, on the contrary, has been reported to have hepatoprotective effects probably through antioxidant, anti-inflammatory, and antiapoptotic effects 13 ; hence, it is unlikely to have contributed to the liver failure.
The dog in the current case report presented initially with gastrointestinal signs but also mild neurologic signs in form of ataxia. It is unclear if the neurologic signs were caused by the hypernatremia, leading to cerebral water loss, the hypovolemia with dehydration, or if the Mn intoxication caused observed ataxia. Neurologic manifestation of Mn intoxication (manganism) has only been reported after chronic exposure in human beings.2,4,18 However, as the ataxia resolved with the resolution of hypovolemia, dehydration, and hypernatremia, it is safe to assume that the neurologic signs were not caused by acute Mn intoxication, unless fluid therapy dropped Mn below levels that would cause neurologic disease. Progression to fulminant hepatic failure eventually led to euthanasia of the dog. It is also unclear if the dog would have recovered from the intoxication with accompanying liver failure by means of ongoing supportive care, as the decision to euthanize was based on the need of further intensive care and severe financial constraints of the owner. Histology found evidence of early hepatocyte and renal epithelial cell regeneration but it is difficult to tell whether this animal would have recovered from the toxic insult without long-term liver disease.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) declare that they received no financial support for their research and/or authorship of this article.