
Abstract
A new commercially available antigen-capture, blocking enzyme-linked immunosorbent assay (antigen-capture b-ELISA), based on baculovirus truncated-S recombinant protein of Transmissible gastroenteritis virus (TGEV) and 3 specific monoclonal antibodies, was developed and evaluated by examining a panel of 453 positive Porcine respiratory coronavirus (PRCoV), 31 positive TGEV, and 126 negative field sera by using another commercially available differential coronavirus b-ELISA as the reference technique to differentiate TGEV- from PRCoV-induced antibodies. The recombinant S protein-based ELISA appeared to be 100% sensitive for TGEV and PRCoV detection and highly specific for TGEV and PRCoV detection (100% and 92.06%, respectively), when qualitative results (positive or negative) were compared with those of the reference technique. In variability experiments, the ELISA gave consistent results when the same serum was evaluated on different wells and different plates. These results indicated that truncated recombinant S protein is a suitable alternative to the complete virus as antigen in ELISA assays. The use of recombinant S protein as antigen offers great advantages because it is an easy-to-produce, easy-to-standardize, noninfectious antigen that does not require further purification or concentration. Those advantages represent an important improvement for antigen preparation, in comparison with other assays in which an inactivated virus from mammalian cell cultures is used.
Keywords
Introduction
Transmissible gastroenteritis virus (TGEV; family Coronaviridae, genus Coronavirus) causes a highly contagious enteric disease of swine characterized by vomiting, severe watery diarrhea, and a high mortality in piglets less than 2 weeks of age. However, in herds with endemic transmissible gastroenteritis (TGE), the majority of piglets are unaffected, which makes the clinical identification of these endemically infected herds difficult. 32 To detect such carrier animals, a simple and reliable diagnostic method for monitoring the status of TGEV infection in herds was required.
Transmissible gastroenteritis virus was first isolated in 1946. 8,10 In 1984, a nonenteropathogenic virus related to TGEV, Porcine respiratory coronavirus (PRCoV), appeared in Europe 27 and later in North America. 47 In contrast to TGEV, PRCoV causes a mild subclinical respiratory infection. 27,47 Until recently, a virus neutralization (VN) assay was used to evaluate antibodies to TGEV. However, antibodies to TGEV and PRCoV cannot be distinguished by using classic methods such as VN, indirect enzyme-linked immunosorbent assay (ELISA), or fluorescent antibody tests, 2,13 which constitutes an important problem for the diagnosis of the disease. 40,42,45,47
Three major structural proteins were described for coronaviruses: spike glycoprotein (S; 180–200 kDa), membrane (M; 21–30 kDa), and nucleoprotein (N; 45–50 kDa) 20,23,30,31,46 (Fig. 1), the S protein being the most interesting from the antigenic and immunogenic points of view. 9,22,41,42 Four antigenic sites, mapped on the S protein in the order C, B, D, and A, starting from the N-terminal end, were recognized on the S protein. 6,7,11,14 Antibodies to those antigenic sites can be found in the serum of TGEV-infected pigs. The absence of 2 of these antigenic sites (B and C) in the S protein of PRCoV as a consequence of the deletion of 224–227 amino acids, was the basis for their differentiation from the enteric viruses. 28,29,33,34 A number of serologic ELISAs based on monoclonal antibodies (mAb) were developed to differentiate TGEV and PRCoV. 1,3,18,35,37,43 However, to the authors’ knowledge, to date, there have been only 2
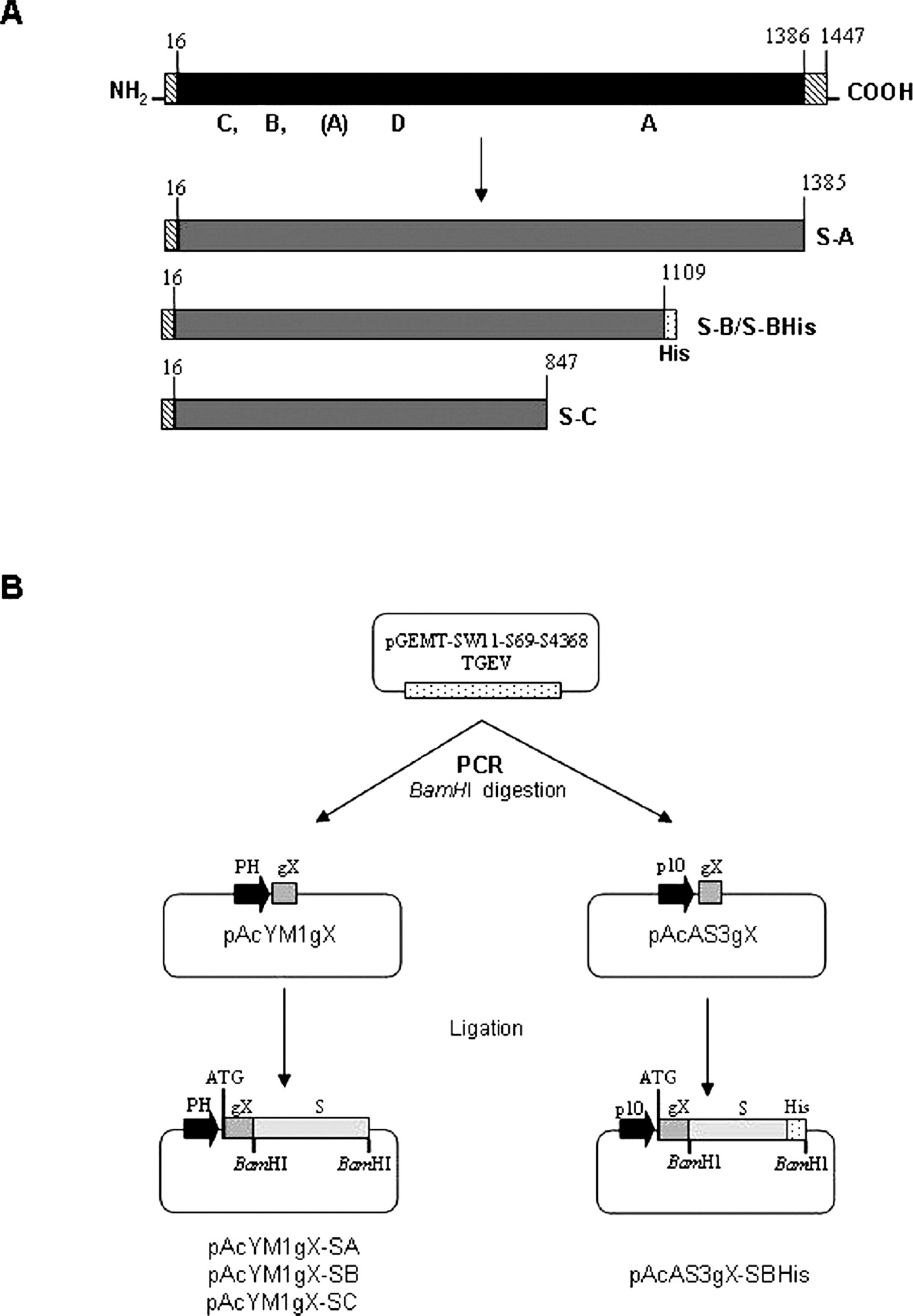
Diagram of complete and truncated forms of Transmissible gastroenteritis virus (TGEV) S protein (panel A) and schematic representation of the construction of baculovirus expression transfer vectors that contain truncated forms of the TGEV S gene (panel B).
commercially available differential assays a , b for use in diagnostic laboratories. 4 Both are based on a blocking ELISA (b-ELISA) and use of a noninfectious semipurified virus or an extract of infected cells as antigen. The use of a recombinant antigen instead of viral antigen in the ELISA is expected to greatly improve the assay because of the high quality of the antigen. Moreover, its use should also allow researchers to avoid some disadvantages of the large-scale preparation of native antigen, such as the lack of standardization of the productive processes, batch variation, low yield, and high production costs. In addition, such antigen preparations may still be infectious and must be handled accordingly.
Baculoviruses are widely used vectors, because recombinant proteins are expressed in large amounts in infected insect cells. Several fragments of the S protein were cloned into the baculovirus transfer vectors to determine if the expressed S protein would be suitable as antigen in a b-ELISA for detection of serum antibody to either TGEV or PRCoV. The authors report the use of a truncated form of the recombinant S protein, released into the cell culture supernatant of baculovirus-infected cells, in an antigen-capture b-ELISA c that detects serum antibody to either porcine coronavirus. The results were compared with a commercially available differential coronavirus b-ELISA. a The antigen-capture b-ELISA offers the advantage of using easily produced noninfectious antigen directly from baculovirus-infected Sf9 cell culture supernatant, without further purification or concentration.
Materials and methods
Cells and viruses
Spodoptera frugiperda insect cell line Sf9, d adapted to grow in medium without serum, was used to propagate recombinant baculoviruses. The cells were grown in suspension or monolayer culture as previously described. 17 Autographa californica multiple nucleopolyhedrovirus (AcMNPV) wild type e was used to purify DNA for the transfections based on the p10 promoter. Recombinant baculoviruses AcYM1gX-SA, AcYM1gX-SB, AcYM1gX-SC, and AcAS3gX-SB-His, which express the TGEV S protein-fragment A, B, or C under polyhedrin or p10 gene promoters, were used for antigen preparation.
Monoclonal antibodies and serum samples
Three S protein-specific mAbs (6AA6, 6AC3, and 1DB12) were used to develop the ELISA assay. Monoclonal antibody 6AC3 recognizes the antigenic site A. Monoclonal antibodies 1DB12 and 6AA6 recognize the antigenic sites B and C, respectively, which are absent in PRCoV. The localization of these sites was performed by testing the antigenicity of proteins fragments and prokaryotic expression products of S gene and by sequencing of mAb-resistant mutants. 6,12,14,34 The anti-S protein mAbs 2BC2 and 5BH1 were used for Western blot assays. All the mAbs were obtained by license agreement with Consejo Superior de Investigaciones Científicas, Spain. A total of 610 sera collected from individual pigs of various ages were used. Of the total sera, 453 were PRCoV positive, 31 were TGEV positive, and 126 were negative, with a blocking commercial test a used as the reference technique. In addition, 8 porcine sera samples, which were positive to Porcine reproductive and respiratory syndrome (PRRS), 8 porcine sera positive to Porcine parvovirus (PPV), 3 sera positive to Porcine circovirus-2 (PCV-2), 3 sera positive to porcine influenza, and 1 serum sample positive to African swine fever virus (ASFV), obtained from experimentally infected pigs, were also analyzed.
Cloning of gX signal sequence in 2 different baculovirus expression vectors
To allow the efficient secretion of the protein to the extracellular medium, the signal sequence of the gene gX of the Suid herpesvirus 1 (alternatively known as Pseudorabies virus [PRV]) 16 was cloned into the baculovirus expression vectors pAcYM1 26 and pAcAS3. 39,44 Two overlapping oligonucleotides gX-forward (5‘-GTTACATCGAT A GATCTCAACAATGAAGTGGGCAACGTGGATTCT CGCCC-3’) and gX-reverse (5‘-AGATTGGATCCGGC CACGACGGTGCGGACCACGAGGAGCCCAGGGCGA-3’) were designed to regenerate the sequence by PCR. The restriction sites BglII and BamHI (in bold), and the initiation codon (underlined) were included in these oligonucleotides. A PCR amplification reaction was carried out in a total volume of 50 μl with 1 U of DNA polymerase, f 200 μmol of each deoxyribonucleotide triphosphate, and 400 ng of each primer. Amplification involved 30 cycles of denaturalization at 94°C for 30 sec, primer annealing at 45°C for 30 sec, and extension at 72°C for 30 sec. A final polishing step was carried out at 72°C for 10 min. The PCR product was digested with BglII and BamHI and cloned directly into BamHI-digested pAcYM1 and pAcAS3 vectors. The new vectors were designated pAcYM1-gX and pAcAS3-gX. All sequences were confirmed with an automatic DNA sequencer. g
Cloning of 3‘-truncated S gene in baculovirus expression vectors
Three carboxy-terminal deletion mutants were constructed by direct PCR with plasmid pGEMT-SW11-S69-S4368-TGEV h as a template and the oligonucleotide S-N1 or S-N2 as forward primers (Table 1). Oligonucleotides S-CA, S-CB, S-CBHis, and S-CC were used as reverse primers to make the PCR products A, B, and C, respectively, which contained the required deletions (Fig. 1). Oligonucleotides were designed to clone the truncated S gene in frame with the signal sequence gX previously cloned in the baculovirus transfer vectors. The BamHI restriction site and termination codon were introduced in the oligonucleotides. The oligonucleotide S-N1, which contains an initiation codon, was used to amplify the truncated forms S-A, S-B, and S-C cloned in the pAcYM1-gX vector. The oligonucleotide
Transmissible gastroenteritis virus S gene DNA primer names, sequences, and positions, used for the construction of the deletion mutants. *

The restriction site BamHI is in bold, and the initiation and stop codons are underlined.
S-N2 was used to amplify the form S-BHis for its subsequent cloning in the pAcAS3-gX vector. The PCR amplification reactions were carried out under similar conditions as described before. Amplification involved 30 cycles of denaturalization at 94°C for 30 sec, primer annealing at 45°C for 45 sec, and extension at 72°C for 3 min. A final polishing step was carried out at 72°C for 10 min. Fragments with BamHI-compatible ends were ligated into BamHI-digested alkaline phosphatase i -treated pAcYM1-gX or pAcAS3-gX. (In some cases, PCR fragments were first subcloned in a cloning vector. j Data not shown.) The resulting constructions were designated as pAcYM1gX-SA, pAcYM1gX-SB, pAcYM1gX-SC, and pAcAS3-gX-SBHis. All sequences were confirmed with an automatic DNA sequencer. g
Transfection and selection of recombinant baculoviruses
The Sf9 cells were transfected with a mixture of transfer vector (2 μg) and linearized viral DNA k or AcMNPV wild type DNA (500 ng) for pAcYM1- or pAcAS3-derived plasmid, respectively, in the presence of a commercial DNA transfection reagent. 1 After 5–6 days, culture supernatant was plated for baculovirus isolation in the presence of 5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside. m The recombinant viruses were selected by their blue-white phenotypes and subsequently plaque purified before high-titer viral stocks were prepared for each virus. 17
Expression and purification of S protein in the baculovirus system
The Sf9 cells adapted to grow in medium without serum (1–106 cells/ml) were infected at different multiplicities of infection (MOI) from 2–10–2 to 2–10–6 plaque forming units (PFU) per cell. The medium was collected from the third day until the 7th day after infection to analyze the recombinant proteins, and the presence of S protein was analyzed either by immunoblotting with a mixture of S protein-specific mAbs (2BC2 and 5BH1) by using standard procedures or by antigen-capture ELISA (ELISA 1). In the case of SB-His protein, it was highly purified by chromatography with nickel-charged high-density resin. n Briefly, the supernatant culture diluted 1:5 in binding buffer (NaCl 0.5 M, Na2HPO4 20 mmol [pH 7.4]) was applied to a Ni++-charged resin, and, after washing with imidazole 50 mmol, the S protein was eluted with elution buffer (imidazole 500 mmol, NaCl 0.5 M, Na2HPO4 20 mmol [pH 7.4]). Purified protein was stored at −70°C until use. The identity of S protein was confirmed by immunoblotting or ELISA with a commercial anti-His mAb° or with the pool of S-specific mAbs.
ELISA procedures
ELISA 1. An antigen-capture ELISA format was used to detect recombinant TGEV-S protein expressed in the Sf9 cell culture supernatant to normalize the protein concentration batch to batch. Briefly, the wells of microtiter plates p were coated with 1 μg of mAb 6AA6 overnight at 4°C in 0.05 M sodium carbonate buffer (pH 9.6). Washes between consecutive steps were performed with 0.05% Tween 20 in phosphate buffered saline (PBS). The S antigen, directly from cell culture supernatant baculovirus-infected Sf9 cells, and 2-fold dilutions of the supernatant in PBS, was captured for 2 hr at 37°C. The amount of S antigen in the supernatants was determined by measuring the optical density after addition of mAb 6AC3 conjugated with horseradish peroxidase (HPR). Bound antibodies were detected by adding tetramethylbenzidine, 3,3’,5,5’ (TMB-MAX q ) as substrate. The reaction was stopped with 0.5 M H2SO4, and the absorbance was measured at 450 nm in an ELISA reader. r The dilution with an optical density between 1 and 1.3, was selected to used in ELISA 2.
ELISA 2. The detection of serum antibody to either TGEV or PRCoV was determined by antigen-capture b-ELISA. Polystyrene microtiter plates were coated with mAb 6AA6 similarly to the previous ELISA. All steps were performed in a final volume of 100 μl. The selected antigen dilution was captured overnight at 4°C. After the capture of the antigen, the plates were stabilized and blocked for 1 hr at room temperature. Washes between consecutive steps were performed with 0.05% Tween 20 in PBS. Serum samples were diluted 2-fold and added in duplicate to individual wells for 1 hr at 37°C. Swine anti-TGEV or anti-PRCoV antibodies, if present in the test samples, were bound to the captured S protein and blocked the subsequent binding of either HPR-conjugated 6AC3 (which recognizes an epitope present in TGEV and PRCoV) and/ or the specific HRP-labeled TGEV mAb 1DB12 that was added to the respective wells. After incubation for 30 min at 37°C, bound antibodies were detected by adding TMB-MAX p as substrate. If there were no specific coronavirus antibodies in the test serum, then the mAbs bind to the plate, and a strong color is obtained. Conversely, if antibodies were present in the sample, then a colorless reaction is obtained. The diagnostic utility of the antigen-

capture b-ELISA was evaluated in a side-by-side comparison with a commercial TGEV/PRCoV b-ELISA, a according to the instructions supplied in the kit. The negative and positive controls used and the cut-off point established in the new assay were the same as those used in the reference technique.
Data analysis
Analysis of the data was performed by using standard statistical methods with Excel. s Intra- and interplate variability was analyzed by testing a single negative serum with the conjugated mAb 96 times in the same plate and 1 strip per plate in 10% of plates of the final batch, respectively. The results were expressed as coefficient of variation (CV), calculated as the ratio of the standard deviation to the mean–100. Sensitivity, specificity, predictive values, and accuracy for the ELISA test were calculated in relation to the reference technique used. Variables measured included the number of true positives (TP), number of true negatives (TN), number of false positives (FP), and number of false negatives (FN). Sensitivity was calculated as 100–TP/(TP + FN), specificity was calculated as 100–TN/(TN + FP), the positive predictive value was calculated as 100–TP/(TP + FP), and the negative predictive value was calculated as 100–TN/(FN + TN). Test accuracy, the proportion of all tests that gave a correct result, was defined as 100–(TP + TN)/ (total samples).
Results
Cloning, expression, and purification of truncated forms of S protein
Two new baculovirus transfer vectors were constructed by cloning the signal sequence of the gene gX of PRV into the pAcYM1 and pAcAS3 transfer vectors. The new vectors, designated pAcYM1-gX and pAcAS3-gX, respectively, were used to clone different truncated forms of the S gene. By using these vectors, the gene concerned was expressed as a soluble protein under the polyhedrin or p10 promoter, respectively. Three truncated forms of the S gene were generated by PCR with specific forward and reverse primers and cloned into the baculovirus transfer vector pAcYM1-gX (Fig. 1). Subsequently, fragment B with a fusion of 6 His-tag in the 3’ end was cloned in the pAcAS3-gX transfer vector. The plasmids obtained were designated pAcYM1gX-SA, pAcYM1gX-SB, pAcYM1gX-SC, and pAcAS3-gX- SBHis.
The Sf9 cells, adapted to grow in medium without serum, were infected independently with the recombinant baculoviruses, which express the truncated forms of the S protein under polyhedrin promoter at different MOI. One aliquot of each infection was collected at 3–7 days after infection, because the cell cultures showed significant cytopathologic effects. The secreted recombinant proteins present in the culture medium were analyzed after sodium dodecyl sulfate-polyacrylamide gel electrophoresis by immunoblotting analyses with a pool of anti-S protein mAbs (Fig. 2A) or with a commercial anti-His mAb (data not shown). Similar expression results were observed with S-B and S-C truncated forms after 5 days of infection at MOI of 10–4; however, because of the low expression level, the S-A form was not detected by Western blot. This protein was only
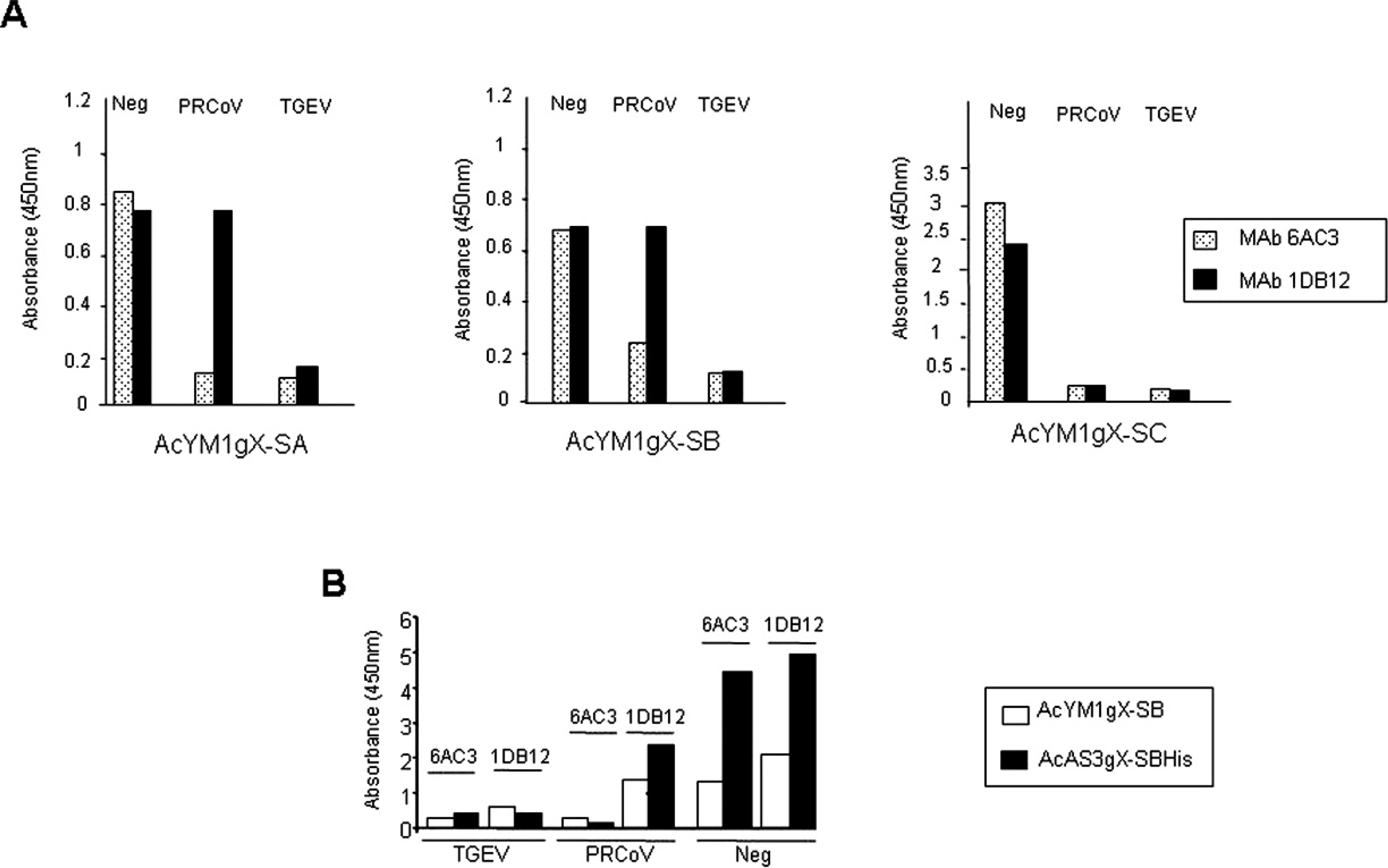
Comparison of truncated forms of Transmissible gastroenteritis virus (TGEV) S protein in an antigen-capture blocking enzyme-linked immunosorbent assay antigen-capture (b-ELISA) for detection of antibodies to TGEV and Porcine respiratory coronavirus (PRCoV).
detected by ELISA after concentration with 65% ammonium sulfate (Fig. 3A). With the objective of improving the yield of the recombinant protein S-B, this DNA fragment was also cloned under the control of a different promoter (p10 promoter), after amplification of the gene with oligonucleotides S-N2 and S-CBHis (Table 1). The Sf9 cells were infected in medium without serum, and the expression of the recombinant protein in the culture supernatant was analyzed by immunoblotting and compared with the expression of AcYM1gX-SB (Fig. 2B). The results showed that the expression of protein AcAS3gX-SBHis under p10 promoter was higher than under polyhedrin promoter. The S-BHis protein was purified by Ni+-conjugated column. Proteins present in the different fractions were analyzed by ELISA and Western blot, the highest concentration being obtained after elution with imidazole 50 mmol (data not shown). However, the final yield of purified S-BHis protein by using this procedure was not high enough, and therefore, it was not used for the development of the new ELISA.
The above results were confirmed by measuring the relative amount of expressed recombinant proteins in baculovirus-infected Sf9 supernatant fluids by an antigen-capture ELISA (ELISA 1, see Materials and methods section). Two-fold dilutions (1:5–1:40) for each time point (48, 72, 96, 120, and 148 hr) were tested for the relative amounts of baculovirus-expressed antigen released into the supernatant fluids. The release of the recombinant protein into the cell culture supernatant fluids was higher with S-BHis than with S-B, and reached a plateau between 96 and 120 hr after infection (data not shown). However, in some cultures, the concentration of the protein decreased very fast at 148 hr. In the plateau region, the cell culture supernatant could be diluted 1:20 without any significant loss of signal.
Incorporation of the expressed SB-His into the antigen-capture b-ELISA
Initially, the 3 truncated forms of the S protein expressed under polyhedrin promoter were evaluated in a standard b-ELISA format by directly adsorbing the antigen from the supernatant of infected Sf9 cells to the wells of 96-well plates. However, this did not result in an adequate signal in the test. The use of concentrated antigen from culture supernatant did not significantly improve the signal. Therefore, an initial antigen-capture step, in which baculovirus-expressed proteins in the culture supernatant were trapped by using the mAb 6AA6 coated to the microtiter plate wells, was incorporated into the assay. The ELISA was initially developed by using the same control sera (negative serum, TGEV-positive serum, and PRCoV-positive serum) that were used in the reference assay. Optimal concentrations of the antigen, HPR-conjugate 6AC3 and 1DB12 mAbs, and substrate solutions were determined in a series of checkerboard titrations of each reagent against all other reagents. Final ELISA conditions were as described in the materials and methods section (ELISA 2). The results, shown in Figure 3A, indicated that only the truncated forms S-A and S-B were possible candidates to be used in the antigen-capture b-ELISA. Both proteins were antigenically similar, and it was possible to distinguish between the PRCoV- and TGEV-positive control sera. Thus, when the absorbance obtained with the negative sera was compared with that obtained with the TGEV-positive sera, blocking percentages of approximately 87.5% (from 0.8 to 0.15 absorbance units) and 85.7% (from 0.7 to 0.1 absorbance units) were obtained with the mAb 6AC3 and the 2 truncated forms, S-A and S-B, respectively. With the mAb 1DB12, the blocking percentage was 75% with form S-A and 85.7% with form S-B, respectively. In the case of PRCoV sera, blocking percentages of 85% and 68% were observed with mAb 6AC3 and with forms S-A and S-B, respectively, and, as expected, no blocking effect was detected with mAb 1DB12. However, when the truncated form S-C was used, the same results were observed with positive PRCoV and TGEV sera, and, therefore, it was not possible to differentiate both sera. The results indicated that the recombinant proteins AcYM1gX-A and AcYM1gX-B were antigenically active, similar to the wild viral protein, and, therefore, suitable for use as antigens to develop an effective diagnostic method that allows for the detection of TGEV and PRCV infections in pigs. However, taking into consideration that the yield data with the truncated form S-A were not as satisfactory as that obtained with form S-B, and that detection of form S-A by ELISA required a concentration of 65% ammonium sulfate, S-B protein was used to continue with the development of the assay. Once the recombinant protein S-B was confirmed as the best recombinant protein, the expression of S-BHis under the p10 promoter was carried out, and its use as antigen in the antigen-capture b-ELISA was also evaluated. The results, shown in Figure 3B, indicated that, when using the more highly concentrated S-B and S-BHis proteins from cell culture supernatant, the blocking percentage was higher when S-BHis recombinant protein was used as antigen compared with S-B. Thus, the blocking percentages of PRCoV sera to the binding of mAb 6AC3 with respect to mAb 1DB12 were 87.5% and 70% with S-BHis and S-B, respectively. The data demonstrated that protein S-BHis produced in the baculovirus system under the control of the p10 promoter could be used as antigen to develop porcine coronavirus diagnostic methods.
Diagnostic utility of the antigen-capture b-ELISA
The reliability and diagnostic utility of the newly developed antigen-capture b-ELISA for detecting and differentiating antibodies against TGEV and PRCoV in several sets of well-defined sera were evaluated by comparison with a commercially available b-ELISA a as a reference test. A total of 453 PRCoV- and 31 TGEV-positive sera from pigs naturally infected with the corresponding virus and 126 negative serum samples were tested. Of the 610 sera analyzed, 600 yielded identical qualitative results (positive or negative) in both tests (reference b-ELISA and antigen-capture b-ELISAs). These data corresponded to an accuracy value of 98.36% (Table 2). The 10 sera with discordant results were negative sera very close to the cut-off value, which seroconverted in PRCoV positives by using the antigen-capture b-ELISA. With this panel of well-characterized sera, the antigen-capture b-ELISA showed a sensitivity and specificity of 100% with the specific TGEV conjugated mAb and a high sensitivity and specificity, 100% and 92.06%, respectively, with the specific coronavirus conjugated mAb.
Intra- and interplate variability was measured in 7 different batches of 300 plates. When intra-plate variability was measured by testing a single negative serum sample 96 times in the same plate, a CV of 4.8% was obtained. For interplate variability, the same serum tested 8 times in 10% of all plates yielded a CV of 4.3%. In both cases, a CV lower than 10% was acceptable. 21 The cross-reaction of the antigen-capture b-ELISA with specific sera from pigs experimentally infected with PRRS, PPV, PCV-2, ASFV, and porcine influenza were examined. As shown in Table 3, no cross-reactivity was observed in the ELISA. The data suggests that the antigen-capture b-ELISA could be used for differentiation of TGEV- and/or PRCoV-infected herds.
Discussion
Several methods have been described to differentiate TGEV from PRCoV infections 1,3,4,15,19,36–38 based on antigenic or genomic differences between TGEV and PRCoV S protein or gene, respectively. Blocking
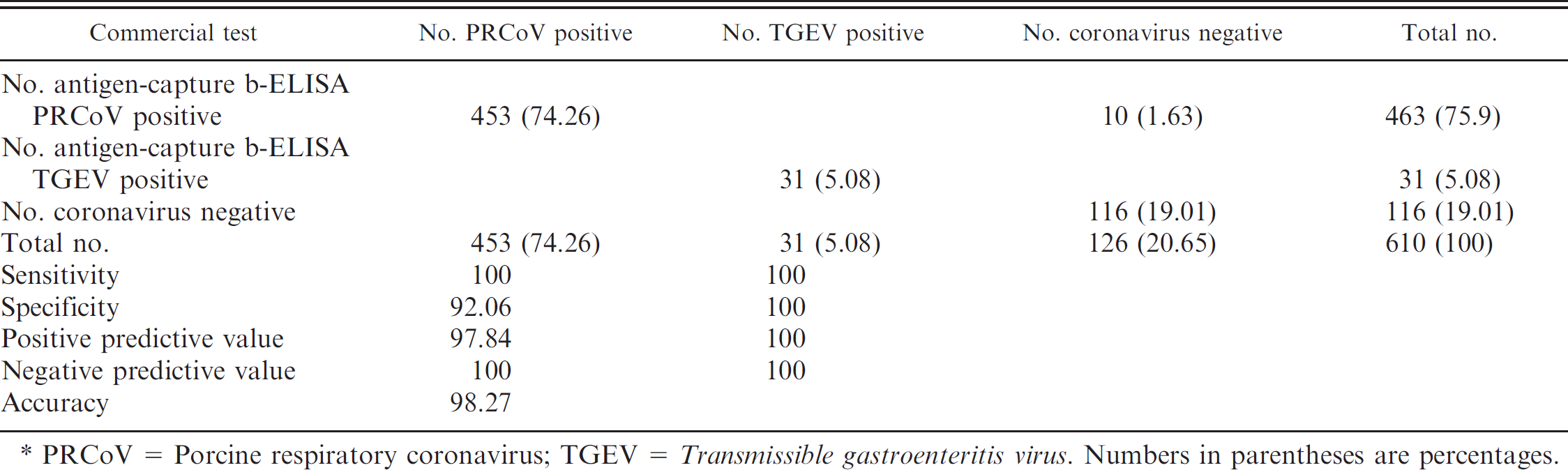
PRCoV = Porcine respiratory coronavirus; TGEV = Transmissible gastroenteritis virus. Numbers in parentheses are percentages.
and competition ELISAs have been used by different investigators with the TGEV S protein as the coating antigen, either from the whole virus or recombinant. However, until very recently, none of the commercially available assays used the recombinant S protein. The purpose of the current study was to evaluate the use of a recent commercially available assay based on a truncated form of TGEV S-protein expressed in the baculovirus system as a potential diagnostic reagent in a b-ELISA to differentiate TGEV- from PRCoV-induced antibodies. The results were compared with a commercial b-ELISA a based on the whole TGEV produced in culture cells. Preparation of cell culture-derived viral antigen for these tests is laborious and variable from batch to batch, and the resulting antigen may be infectious. However, the baculovirus system has been extensively used to express large quantities of proteins that are antigenically similar to their native counterparts and can be used in standardized assays to provide consistent results. In fact, this system has been widely used to produce recombinant proteins for use in diagnostic assays. 5,24,25 In the present study, initially, 3 different fragments of the S gene of TGEV were cloned and expressed under polyhedrin promoter by using the baculovirus expression system. In all cases, DNA fragments were cloned, without the transmembrane region, in frame with the leader sequence gX, which allows the efficient secretion of the protein to the extracellular medium. However, the yield of the biggest fragment (fragment S-A) was very low, and it was not possible to distinguish between TGEV and PRCoV. The best results were observed with fragment B, probably because of its intermediate size, which is antigenically similar to the viral S protein. This recombinant protein was successfully used in antigen-capture b-ELISA for detection of serum antibodies to TGEV and PRCoV. The antigen-capture b-ELISA reported in the current study incorporates an antigen-capture step in a blocking format. The addition of this step allowed the use of baculovirus-infected Sf9 culture supernatant fluid directly without additional purification or concentration. Attempts to improve the test by first purifying or concentrating the recombinant protein from the supernatant fluids did not produce good results while
Cross-reactivity of swine sera against other viruses in antigen-capture blocking enzyme-linked immunosorbent assay antigen-capture (b-ELISA). *
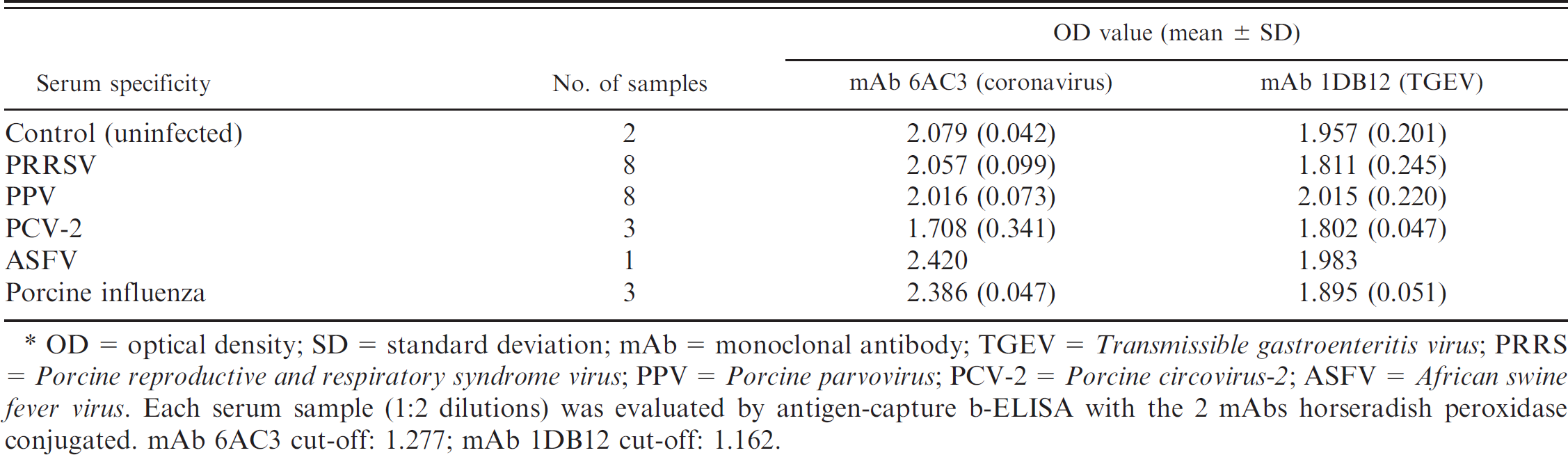
OD = optical density; SD = standard deviation; mAb = monoclonal antibody; TGEV = Transmissible gastroenteritis virus; PRRS = Porcine reproductive and respiratory syndrome virus; PPV = Porcine parvovirus; PCV-2 = Porcine circovirus-2; ASFV = African swine fever virus. Each serum sample (1:2 dilutions) was evaluated by antigen-capture b-ELISA with the 2 mAbs horseradish peroxidase conjugated. mAb 6AC3 cut-off: 1.277; mAb 1DB12 cut-off: 1.162.
adding significant time and labor to the procedure. Binding of expressed antigen directly to the wells of the microtiter plate may result in partial denaturation of the epitopes and inhibit binding of the serum antibodies; however, capturing the antigen with a mAb may help to preserve the antigenic integrity of the epitopes and facilitate the binding of the serum antibodies.
The advantage of the antigen-capture b-ELISA based on recombinant protein over standard b-ELISA based on viral antigen is mainly related to antigen production. Several liters of cell culture supernatant from recombinant baculovirus-infected Sf9 cells can be easily produced in a few days. Once the material is harvested and the titer of antigen is determined, this material can be used directly, without further purification, as a source of noninfectious antigen in the antigen-capture b-ELISA. This fact is very important in a production process because of the simplification of purification procedures and product cost reductions. Another advantage of using a recombinant protein as antigen in the ELISA is that it is possible to avoid the infection of culture cells with the virus and its purification, which is time consuming and expensive.
Monoclonal antibody 1DB12 recognizes the antigenic site B, present in TGEV but not in PRCoV. It could be asked why the absorbance value obtained with this mAb in a blocking assay with a negative serum is higher than with a PRCoV-positive serum (Fig. 3B), if neither has antibodies able to block the binding of the 1DB12 mAb. In PRCoV-positive sera, the presence of other antibodies capable of binding to the recombinant protein could be enough to produce some steric effect partially blocking the binding of mAb 1DB12. However, this fact did not represent any difficulty, and ultimately, the ELISA had a 100% sensitivity for the detection of antibodies to TGEV and PRCoV and 100% and 92.06% specificity, respectively. However, the accuracy was higher than 98%, which indicated that the ELISA could be used as a differential diagnosis technique. To the authors’ knowledge, this is the first report in which a commercially available kit based on recombinant proteins expressed in the baculovirus system for the differential serologic diagnosis of TGEV and/or PRCoV was developed and evaluated.
Acknowledgements
The authors thank Prof. Luis Enjuanes (National Center of Biotechnology) for the construction of pGEMT-SW11-S69-S4368 TGEV. This study was partially supported by a grant (Ref: IDE2001–0695) from the Ministerio de Ciencia y Tecnologí (Spain).
Footnotes
a.
INGEZIM Differential coronavirus, Inmunología y Genetica Aplicada S.A., Madrid, Spain.
b.
Svanova Biotech AB, Uppsala, Sweden.
c.
INGEZIM Differential coronavirus 2.0, Inmunología y Genetica Aplicada S.A., Madrid, Spain.
d.
Cell line Sf9, CRL1711, American Type Culture Collection, Barcelona, Spain.
e.
Gift from Dr. Summers, Texas A&M University, College Station, TX.
f.
VentRr DNA polymerase, New England Biolabs Inc., Ipswich, MA.
g.
ABI PRISMr 3130 DNA automatic sequencer, Applied Biosystems, Foster City, CA.
h.
Kindly donated by Prof. Enjuanes, Centro Nacional de Biotecnología (CNB), Madrid, Spain.
i.
Alkaline phosphatase, Roche Diagnostic, Barcelona, Spain.
j.
pGEMr–T Easy Vector System, Promega Biotech Ibérica SL, Madrid, Spai.
k.
BacPAK6 Viral DNA, Clontech Laboratories Inc., Mountain View, CA.
l.
Jet PEITM DNA Transfection Reagent, Polyplus-transfection SA, Illkirch, France.
m.
X-Gal (5–bromo–4–chloro–3–indolyl-β-D-galactopyranoside), Fermentas International Inc., Burlington, Ontario, Canada.
n.
His-Bind Resin, Hispanagar SA, Burgos, Spain.
o.
Anti-His MAb, Sigma-Aldrich química S.A, Madrid, Spain.
p.
Polystyrene microtitre plates, Lab Systems, Barcelona, Spain.
q.
TMB (MAX), Neogen, Lansing, MI.
r.
Multiskan Ascentr, Thermo Fisher Scientific Inc., Waltham, MA.
s.
Excelr, Microsoft Corp., Silicon Valley, CA.