
Abstract
Invertebrate iridoviruses (IIV) have been a regular problem for insect breeders. They have also recently been isolated from various lizard species. An iridovirus isolated from several tissues of a high-casqued chameleon (Chamaeleo hoehnelii) was identified as an IIV on the basis of electron microscopy (EM), sequencing of a portion of the major capsid protein (MCP) gene, and restriction endonuclease analysis of viral DNA. The pathogenicity of this isolate for crickets of the species Gryllus bimaculatus was tested by using 3 experimental infection studies. The mortality rates in the infected crickets ranged between 20% and 35%. The fat bodies of the crickets were examined on cell culture, with a nested PCR targeting the MCP gene, histologically, with in situ hybridization and by EM. Nested PCR was the most sensitive method for detecting IIV in the fat-body samples. Virus was re-isolated from several fat-body samples. In some fat bodies of infected crickets, massive arrays of viruses could be detected by EM. These findings support the hypothesis that IIV from insects are able to infect reptiles.
Introduction
Iridoviruses are 120 to 300 nm in diameter, with a double-stranded DNA genome and an icosahedral capsid that contains a lipid component. The family Iridoviridae is currently divided into 5 genera: Iridovirus, Chloriridovirus, Ranavirus, Lymphocystivirus, and Megalocytivirus. 1 Until recently, viruses of the genera Iridovirus and Chloriridovirus had only been described in invertebrates. They are of particular interest because of their potential use for controlling important agricultural pests and vector insect species in which they induce lethal infections. 6 Ranaviruses are known to infect fish, amphibians, and reptiles; lymphocysti-viruses and megalocytiviruses occur in fish.
At the end of the 1990s, 2 research groups in Germany isolated and characterized iridoviruses from crickets (Orthoptera, Gryllidae) of the species Gryllus campestris and Acheta domesticus 6 and Gryllus bimaculatus. 5 In both cases, the insects originated from commercial breeders who produced crickets as food for vertebrates, especially reptiles. The isolated viruses were named cricket iridovirus (CrIV) 6 and G. bimaculatus virus (GbIV). 5 Both were found to be related to IIV-6, the type species of the genus Iridovirus. In infection studies on the host range of CrIV, this isolate was successfully transmitted to other Orthopteran species, which included G. bimaculatus. 3
Insect iridoviruses belonging to the genus Iridovirus have only recently been described from reptilian hosts. In 2001, a German group reported the isolation of IIV-like viruses from 2 bearded dragons (Pogona vitticeps), a chameleon (Chamaeleo quadricornis), and a frilled lizard (Chlamydosaurus kingii). PCR amplification of purified viral DNA with primers that correspond to a portion of the gene encoding the major capsid protein (MCP) of IIV-6 amplified products with 97% identity to the nucleotide sequence of IIV-6 and 100% identity to the nucleotide sequence of GbIV. A host-switch of this virus from prey insects to the predator lizards was postulated. 4
After isolating an IIV from a high-casqued chameleon, transmission studies were carried out with crickets to see if a virus isolate from a lizard was pathogenic for an invertebrate species. In the course of these studies, diagnostic tests, including a nested PCR and in situ hybridization (ISH) for the detection of IIV in insects and lizards were developed and compared.
Materials and methods
Virus isolation
A juvenile high-casqued chameleon (Chamaeleo hoehnelii) died several weeks after purchase from a wholesale dealer. On postmortem examination, the animal was cachectic. A keratoconjunctivitis was noted. The liver was slightly hyperemic, and the spleen was slightly enlarged. The gastro-intestinal tract was empty. Lung, large and small intestine, liver, kidney, and spleen were collected for virus isolation. Tissues were placed in basal medium Eagle (BME) with Earle's salt solution with 2 mM L-glutamin, 200 U/ml penicillin G, 380 U/ml streptomycin sulfate, 6.4 U/ml gentamycin sulfate, and 0.5 μg/ml amphotericin B. a Samples were sonified (Branson Sonifier 250, b 3 pulses at 30%), centrifuged at low speed for removal of cell detritus, and inoculated onto 2 reptilian cell lines: Terrapene heart cells (TH-1) c and Viper heart cells (VH2) c for virus isolation. TH-1 and VH2 were cultured with BME with 2% fetal calf serum (FCS). Tissue cultures were incubated at 28°C with 5% CO2.
Fat-body samples from infected crickets were also inoculated onto TH-1 and VH2 cultured with BME or Dulbecco's modified Eagle Medium (DMEM) a with 2% FCS. Fat-body samples of all crickets were collected in BME with double the amount of antibiotics. The samples were sonified, centrifuged and, in some cases, filtered with 0.45-μm filters. d
Cell cultures were observed daily for cytopathic effects (CPE). All cultures were harvested 3 to 5 days after the first appearance of CPE. Identification of the virus isolated from the chameleon was carried out by electron microscopy (EM) followed by restriction enzyme digestion of viral DNA with the enzymes MspI and HpaII as previously described 7 ; MspI and HpaII are isoschizomers. Both recognize the sequence CCGG, but HpaII does not digest DNA if the internal cytosine is methylated, as is the case for ranaviruses. A PCR targeting the MCP gene of IIV was carried out with DNA from the isolated virus as previously described. 4 A 510-bp amplicon was sequenced as described below. To determine whether this putative IIV from a lizard was truly infective for insects, a series of transmission studies were carried out with crickets (G. bimaculatus).
Insects
An iridovirus-negative colony of G. bimaculatus was established by using crickets obtained from another institution. e The crickets had no contact to other insects, were fed with lettuce and dog food, f and regularly tested negative for IIV by nested PCR.
Bioassays
To study the virulence of an IIV from a chameleon in invertebrates, per os transmission experiments were carried out as previously described 6 with third instars of G. bimaculatus. A total of 20 nymphs were completely immersed in cell-culture supernatant from infected TH-1 cells with a virus titer of approximately 106.5 TCID50/ml for approximately 15 sec. Ten nymphs were dipped into TH-1 supernatant as negative controls. In view of the cannibalistic behavior of this species, all insects were housed individually in beakers (diameter, 8.5 cm; height, 9.5 cm). The insects were kept at room temperature. The studies lasted 71 (first transmission study) and 50 days (second and third transmission studies), and mortality was recorded daily. Environmental temperatures were measured regularly with a minimum-maximum thermometer. Fat-body samples were collected from all dead crickets and prepared for virologic, histologic, ISH, and EM investigations. All crickets remaining alive at the end of each study were euthanized by decapitation and were also tested.
Extraction of viral DNA
Fat bodies from all crickets were collected in BME as described for virus isolation. A DNeasy tissue kit g was used for DNA preparation from 200 μl of supernatant from this material according to the manufacturer's protocol for the isolation of DNA from animal tissues. The prepared DNA was eluted in 100 μl buffer AE.
Nested PCR and sequencing of PCR products
First, a PCR that amplifies a 510-bp region of the MCP gene with the primers PCRFOR 5′-CCATTACATT-TAATGATTTGG-3′ and PCRREV 5′-TTTTGACGTG-GTGCAGTTTGAAC-3′ 8 was used. An aliquot of 1 μl tissue supernatant in BME was added to 49 μl PCR solution, containing 1.5 mM MgCL2, 350 μM (dNTP), 50 pmol of each primer and 1.25 U Taq polymerase h in Taq buffer. The PCR was carried out as previously described. 8
Because in some cases in which virus isolation was successful the PCR delivered negative results, a nested PCR was developed. In a comparison, the nested PCR was 100 times more sensitive than the PCR without the nested step and 10 times more sensitive than cell culture. The nested PCR was used for amplification of an approximately 270-bp long part of the genomic region encoding the MCP. The outer primers MCP-F1 (5′-GGTTTCATCGATATCGCCAC-3′) and MCP-R4 (5′-GAAAAGTAATCACTGCCCAT-3′) and the inner primers MCP-R2 (5′-AGCAGAAACATTTCCAATCAT-3′) and MCP-F3 (5′-GGGCCGGAGATTATTTGTT-3′) were previously described. 3 For first round PCR, 5 μl of extracted DNA were included in 50 μl reactions that contained 2.5 mM MgCl2, 350 μM of each dNTP, 40 pmol of each primer, and 1.25 U Taq DNA polymerase h in Taq buffer. For the second round, 1 μl of the first round product was included in 50 μl reactions. Before amplification, the reaction mixtures were incubated at 96°C for 5 min. Amplification conditions were 35 cycles of denaturation at 95°C for 45 sec, annealing at 50°C for 45 sec, and extension at 72°C for 1.5 min. This was followed by incubation at 72°C for 10 minutes. RNA extracted from the fat bodies of iridovirus-negative crickets bred in our laboratory were used as negative controls in all PCR assays. To determine the specificity of the assay, products from 2 crickets (one only positive by nested PCR, the other also positive by cell culture, histology, and EM) were directly sequenced. The DNA fragment obtained by PCR was separated by agarose gel electrophoresis, purified by using a kit, i and directly sequenced with the PCR primers on an ABI 310 capillary sequencer by using a sequencing kit j after the manufacturer's protocol.
Light microscopy
After the postmortem examination, fat-body samples of selected crickets were fixed for 48 hr in Bouin solution of 714.3 ml/liter picric acid, 238.1 ml/liter formalin (36%), and 47.6 ml/liter acetic acid, k paraffin embedded, cut at 3 μm, and stained with hematoxylin and eosin.
In situ hybridization
Two different digoxigenin (DIG) labeled probes were made for the ISH studies after a modified protocol originally described by Emanuel 2 by using the primers PCRFOR and PCRREV and MCP-F3 and MCP-R2 in the PCR assays described above. In both probe reactions, the dNTP mix was substituted by the prescribed amount of DIG DNA labeling mix. 1 The probes were tested for specificity by Southern blot hybridization against various concentrations of 4 different IIV isolate partial MCP gene PCR amplicons and λ-phage DNA as a negative control.
For the ISH studies, tissues were fixed in Bouin solution and embedded in paraffin. Sections were dewaxed with xylene and rehydrated before proteolysis by proteinase K. After refixation in absolute ethanol, they were air-dried. Hybridization was carried out in a humid chamber at 40°C overnight in the following solution: 2.5 μl herring sperm-DNA, 1 μl probe, and 3.5 μl formamide were mixed and denatured for 5 min at 95°C. On ice, an additional 21.5 μl formamide, 10 μl 20 × SSC solution (3 M NaCl, 0.3 M Nacitrate, pH 7), 1 μl 50 × Denhardt solution and 5 μl 50% dextran sulfate and bidistilled water were added to a final volume of 50 μl. After further washing steps, the specific color reaction was performed during 60 min, and tissues were sealed with aqueous mounting medium. m The tissues were examined by using a light microscope. n
Electron microscopy
Fat body samples from selected crickets were fixed in 2.5% glutaraldehyde o for 2 hr. After fixation, the samples were washed 3 times for 10 min in 0.1 M sodium cacodylate buffer (pH 7.4) 1 and fixed in a 1% solution of osmium tetroxide o in 0.1 M sodium cacodylate buffer for 1 hr at 4°C. A washing step with sodium cacodylate buffer was repeated, and the samples were washed 3 times with 0.1 M acetic acetate buffer (pH 5), p followed by contrasting with a saturated uranyl acetate solution (5%) q in 70% ethanol for 2 hr at room temperature. Dehydration was carried out in ascending concentrations of ethanol (70%, 95%, 100%, 100%, 100%). Propylenoxide r was used as an intermediate solvent, and the samples were then embedded in an epoxy resin. o After 48 hr in an incubator at 60°C, the blocks were polymerized, routinely sectioned, adsorbed onto 300-mesh copper grids, and examined in an electron microscope s at 80 kV.
Results
Virus was isolated from all of the chameleon tissues tested (lung, large and small intestine, liver, kidney, and spleen). The EM of cell-culture supernatant showed icosahedral iridovirus-like particles approximately 100 nm in diameter. The virus was identified as an IIV based on a lack of methylation of the genomic sequence CCGG as indicated by susceptibility of DNA from the isolate to digestion with both MspI and HpaII. A 510-bp amplicon was obtained by using the PCR with the primers MCPFOR and MCPREV as described. Sequencing of the product showed that it was 100% identical to the corresponding sequence in GbIV (GenBank accession no. AF247641).
Results of the diagnostic assays carried out on infected Gryllus bimaculatus in transmission studies I to III.∗
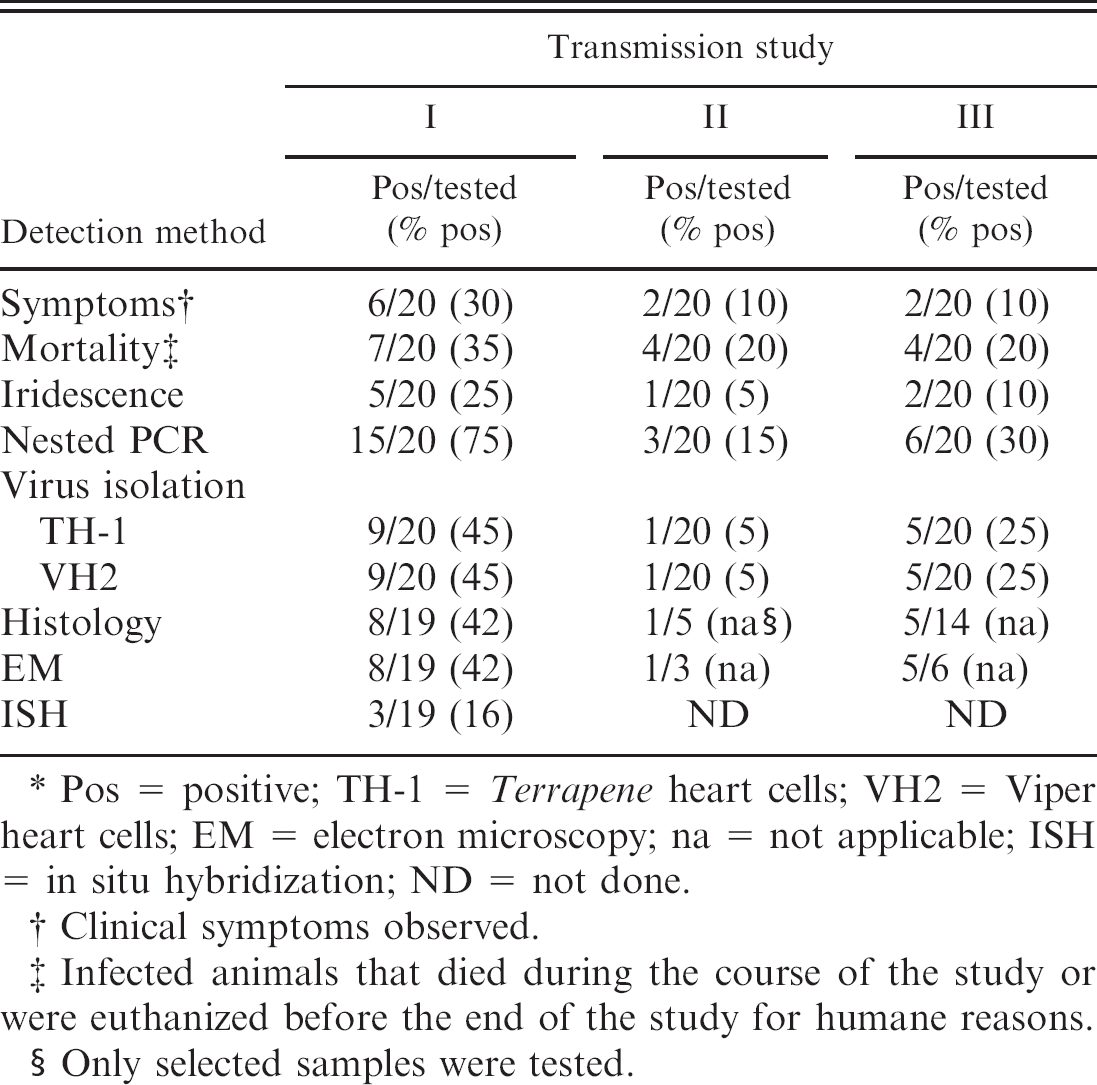
Pos = positive; TH-1 = Terrapene heart cells; VH2 = Viper heart cells; EM = electron microscopy; na = not applicable; ISH = in situ hybridization; ND = not done.
Clinical symptoms observed.
Infected animals that died during the course of the study or were euthanized before the end of the study for humane reasons.
Only selected samples were tested.
In 3 transmission studies, a total of 60 crickets were infected, and 30 crickets were used as controls. All examined control crickets were negative in all assays. In the first transmission study, all 20 infected crickets were subjected to all assays. In transmission studies II and III, some crickets were selected for histology, ISH, and EM. The results of the transmission studies are presented in table 1.
During transmission study I, 6 of 20 infected crickets (30%) died and 1 (5%) was euthanized before the end of the 71-day study. In addition, 1 of the negative controls (5%) died. Some of the infected crickets showed swollen abdomens and distress as major symptoms, and postmortem examinations revealed that 5 of the fat bodies of infected crickets (25%) were bluish iridescent in color. Of these crickets, 3 (15%) died during the study. Altogether, 15 of the infected crickets (75%) were iridovirus positive in at least one of the diagnostic assays used.
During transmission study II, 4 of the infected crickets (20%) and 2 of the controls (10%) died. One of the infected crickets (5%) that died before the end of the 50-day study showed iridescence. A total of 3 of the infected crickets (15%) were iridovirus positive in at least one of the diagnostic assays used.
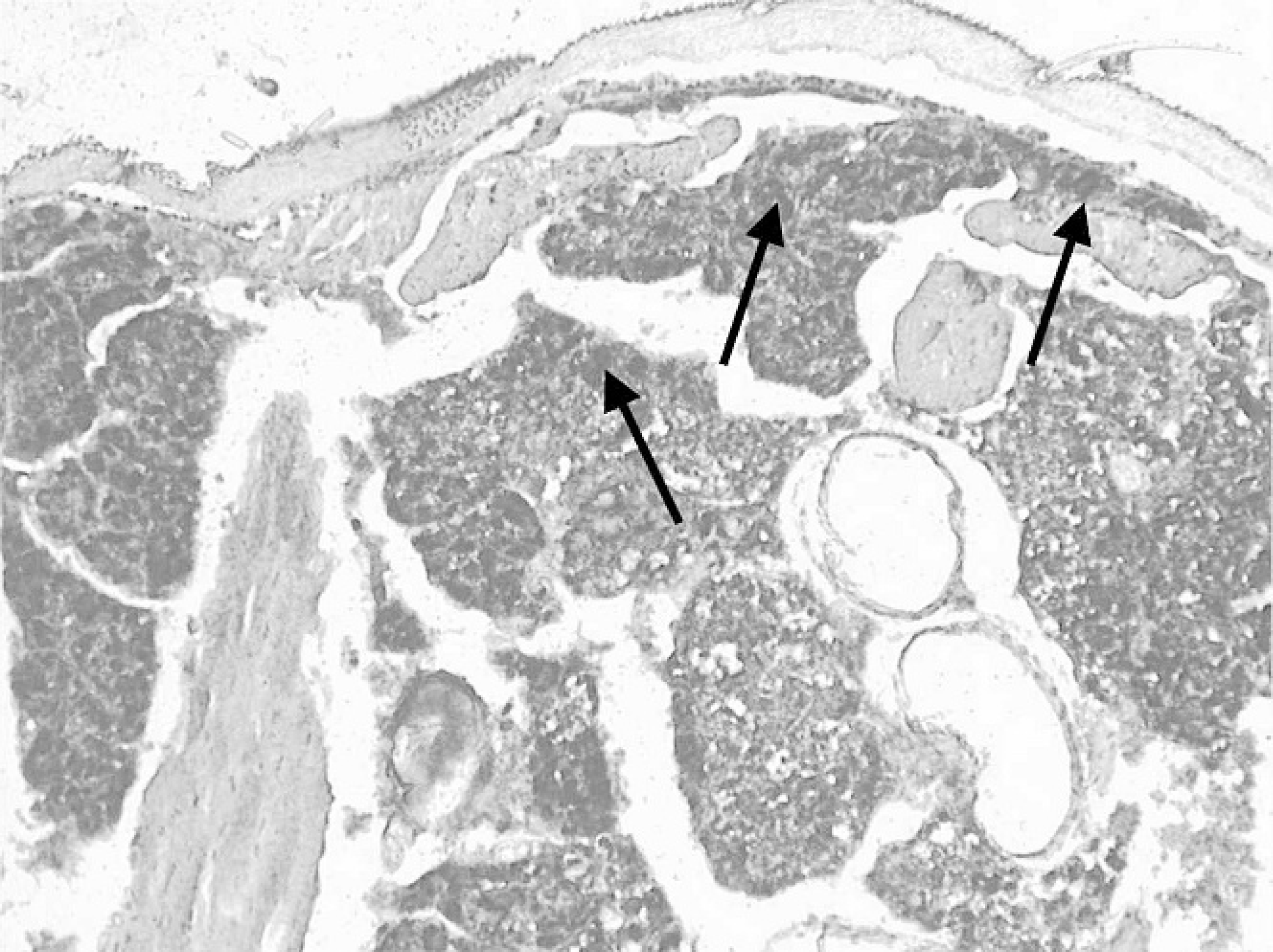
Section of the fat body of a cricket (Gryllus bimaculatus) infected with an invertebrate iridovirus (IIV) isolated from a chameleon. In situ hybridization with digoxigenin labeled IIV major capsid protein gene shows IIV DNA as black deposits in the fat-body tissue (arrows). 20×.
In transmission study III, 3 of the infected crickets (15%) died and 1 (5%) was euthanized before the end of the 50-day study. Two infected crickets (10%) showed iridescence, one of which died during the transmission study. A total of 6 of the infected crickets (30%) were iridovirus positive in at least one of the diagnostic assays used.
Deaths of negative control crickets during the course of the transmission studies were because of egg-laying problems, age, or shedding difficulties. None were associated with virus. Minimum temperatures in the infected groups were 21.0°C (I), 18.9°C (II), and 20.1°C (III). The maximum temperatures were 28.1°C (I), 31.2°C (II), and 29°C (III). Average temperatures were 23.8°C for study I, 25.3°C for study II and 22.7°C for study III. Light microscopy showed a network of dense granular viral stroma in the obviously hypertrophied fat-body cells of some of the infected crickets as previously described. 6 All of the examined negative controls showed normal fat-body tissue. Strong signals were detected in the fat bodies of crickets with large amounts of virus by ISH (Fig. 1). The EM showed massive accumulations of virus particles in the fat-body tissue of some of the infected crickets (Fig. 2).
A statistical calculation with the chi-square test was carried out with all of the results of the nested PCR. This showed that G. bimaculatus could successfully be infected with an IIV isolated from a chameleon, with a probability of error of 0.1%. A statistical comparison of all of the results of the nested PCR by using a chi-square test showed that there is a significant difference between the 3 infection studies. A statistical comparison of cell culture and nested PCR with the McNemar test shows that the nested PCR is the most sensitive method. A comparison of the cell-culture results with the 2 cell-culture media used showed that the total amount of virus isolations was the same. In 7 of 11 cases from which virus was isolated, an isolate was obtained with both media. In addition, 2 isolates were obtained only with BME on TH-1 or DMEM on VH2.
Several statistical comparisons of 2 methods at any one time by the chi-square test showed that, with a probability of error of 0.1% nested PCR, cell culture, and histology give the same results. In the case of EM and ISH, the probability of error was 2.5% compared with the nested PCR. But, in the case of these 2 methods, the number of samples examined was too low for a reliable statistical evaluation. Sequencing of the 2 PCR amplicons of the 2 cricket samples showed that they were 100% identical to the corresponding sequence of the isolate from the high-casqued chameleon (C. hoehnelii).
Discussion
Insect iridoviruses have been repeatedly isolated from lizards in Germany over the past several years. Although it has been hypothesized that these viruses originated in prey insects fed to the lizards, 4 the characterization of the IIV isolates remains incomplete, and this hypothesis has not been proven. The transmission studies with crickets of the species G. bimaculatus and an IIV isolated from a high-casqued chameleon (C. hoehnelii) showed that crickets are susceptible to this lizard isolate.
Infection studies on the host range of the IIV isolate CrIV with various Orthopteran species, including G. bimaculatus, have been described in detail. 6 In the orally infected insects that isolate led to characteristic symptoms and a mortality of 93% after dipping in virus suspension and 48% after feeding virus infected food. The present transmission studies only resulted in mortalities that ranged from 20% to 35% after dipping. Some of the crickets that were positive in the diagnostic assays did not show any symptoms or die during the trial. Three crickets (2 in infection study I, 1 in infection study III) even showed the typical bluish iridescence during dissection at the end of the study, without showing any prior sign of infection. These highly infected, virus-laden but lesion-free insects are most likely a risk for transmission and distribution of IIVs. They can infect stocks of invertebrates when introduced to naive breeding colonies. It is also more difficult to identify and eradicate an iridovirus infection in an invertebrate breeding colony when it is not possible to identify live infected insects. It also seems more likely that clinically inapparent virus carriers with a high virus load might infect vertebrates when fed to insectivorous reptiles.

Electron micrograph of the fat body of a cricket (Gryllus bimaculatus) infected with an invertebrate iridovirus isolated from a chameleon: Assembly site. Large numbers of electron-dense icosahedral viral particles are visible. 8,000×.
The insects from which positive results were obtained with the less sensitive methods used (EM, ISH) were also positive with the more sensitive ones (nested PCR, cell culture), demonstrating a high specificity of the tests used. One exception was infected cricket number 7 of infection study I. In that special case, virus isolation on cell culture was not successful, although histology and EM were positive. The micrographs in the EM showed massive arrays of clearly ordered crystals of viruses, which led to the hypothesis that, in this case, the crystals were so big that they were filtered out of the cell-culture sample with the 0.45-μm filters.
Based on the results of this study, the nested PCR is the most sensitive method for the detection of an iridovirus infection in crickets. However, nested PCR has an increased risk of contamination and is also more costly and time consuming than a single round of PCR. This PCR must be handled very carefully in the laboratory, with steps taken to avoid contamination, especially because many of the tissues examined may contain large amounts of virus.
The significant differences between the 3 infection studies may have been caused by differences in the insects used for the infection study and their age, the aliquot of the isolate used for the infection of the crickets, or the temperature during each trial. All of the crickets used in the infection studies came from the same breeder and were about the same age at the beginning of the 3 trials. For all 3 infections, aliquots of the same passage of the IIV isolate from the high-casqued chameleon (C. hoehnelii) were used. A comparison of the temperatures of all 3 transmission studies showed that the highest absolute minimum and the lowest absolute maximum temperatures were recorded during infection study I (with the highest infection rate). This means that the temperature was relatively stable within a narrow range. In contrast, the lowest absolute minimum and the highest absolute maximum temperatures were recorded during infection study II (with the lowest infection rate), which means the temperature varied the most during this study. The most probable explanation for the significant differences between the infection rates in the individual assays is the temperature differences during the 3 trials. The range of the temperatures may be the main factor in these differences. Iridoviruses have temperature optimums within which they grow best. This leads to the hypothesis that, during an infection study with less variation in temperature, the highest infection rate could be achieved. It would be best to conduct future studies at constant temperatures that should not exceed 28°C.
IIVs have been increasingly detected in lizards over the past few years. The detection of these viruses in lizards has previously been carried out by virus isolation followed by virus identification. These methods are time consuming and expensive. The methods developed here may help to increase the sensitivity and speed of IIV diagnosis. The fact that invertebrate viruses may be transmitted to vertebrates also increases the importance of screening measures for the commercial production of prey insects.
Acknowledgements
This study was financed by the Deutsche Forschungsgemeinschaft. The authors would like to thank Dr. Udo Hetzel, Department of Veterinary Pathology, Faculty of Veterinary Medicine, the University of Liverpool, for postmortem examinations and submission of the chameleon case. The authors thank Silvia Speck, Christa Schäfer, and Gabriele Czerwinski for help and excellent technical support.
Footnotes
a.
Biochrom AG, Berlin, Germany.
b.
Branson Ultrasonics, Danbury, CT.
c.
American Type Culture Collection, Teddington, Middlesex, United Kingdom.
d.
FP 30/0,45 CA-S, Schleicher and Schuell MicroScience, Dassel, Germany.
e.
Dr. Regina Kleespies, Biologische Bundesanstalt für Landund Forstwirtschaft, Darmstadt, Germany.
f.
Matzinger Hundeflocken, Nestlé Purina Pet Care, Euskirchen, Germany.
g.
Qiagen, Hilden, Germany.
h.
Fermentas GmbH, St. Leon-Rot, Germany.
i.
Invisorb Spin DNA Extraction Kit, Invitek GmbH, Berlin, Germany.
j.
ABI PRISM BigDye Terminator v1.1 cycle sequencing kit, PE Applied Biosystems, Foster City, CA.
k.
Merck, Darmstadt, Germany.
l.
Roche Diagnostics GmbH, Mannheim, Germany.
m.
Aquatex, Merck, Darmstadt, Germany.
n.
ZEISS Axioskop 2 with Sony digital camera, Göttingen, Germany.
o.
Plano, Wetzlar, Germany.
p.
Roth, Karlsruhe, Germany.
q.
Fluka, Deisenhofen, Germany.
r.
Serva, Heidelberg, Germany.
s.
JEM-100SX, JeolLTD, Tokyo, Japan.