
Abstract
Background
In Serbia, as well as worldwide, clinical trials are a significant and unavoidable part of the development of new medicines. There is limited research on prospective risk management in clinical trials in Serbia, underscoring the need for systematic approaches, such as the failure mode and effects analysis (FMEA) method. The effectiveness of this complex process largely depends on knowledge and experience in the field of management. Like any other process, clinical trials are subject to various risks for both the company and the patient.
Purpose
The purpose of this article is to conduct a prospective, qualitative study on the risk management of clinical trials in the Republic of Serbia, to identify, assess, and reduce risks for both patients and companies, and to offer corrective measures for reducing their frequency and consequences.
Materials and Methods
A focus group applied the FMEA method to identify, assess, and mitigate risks. Each risk was assessed based on severity, probability, and detectability, and then a risk priority number (RPN) value was calculated. For the most significant risks, corrective measures were proposed, and the RPN was recalculated. The research was conducted from December 12, 2022 to March 8, 2023.
Results
In total, 20 risks were identified with RPN values ranging from 10 to 192, of which five had RPN > 100. The minimum RPN value among the five most significant risks was 105, and the maximum was 192. After the proposed corrective measures, there was a significant reduction in the RPN values across all five risks, ranging from 57.14% to 83.33% when compared to the initial values.
Conclusion
The FMEA method has proven to be a useful instrument not only for identifying and predicting potential risks, but also for their control.
Introduction
Clinical Trials
In literature, the concept of clinical trials is defined in different ways. According to the European Union Directive 2001/20, a clinical trial is
any investigation in human subjects intended to discover or verify the clinical, pharmacological and/or other pharmacodynamic effects of one or more investigational medicinal product(s), and/or to identify any adverse reactions to one or more investigational medicinal product(s) and/or to study absorption, distribution, metabolism, and excretion of one or more investigational medicinal product(s) with the object of ascertaining its (their) safety and/or efficacy.
1
This definition is explained in the new regulation no. 536/2014 of the European Parliament and of the Council, and in order to achieve such objective, it is beneficial to define certain terms in more detail, for example, clinical studies, provided that this term includes clinical trials as well. The aforementioned model relies on international guidelines and expands on the Union regulations on medicines, which distinguish between clinical trials and non-interventional studies. 2 On the other hand, this term can also imply that a “clinical trial is a prospective study comparing the effects and value of intervention(s) against a control in human beings.” 3
According to certain authors, ways to assess the effectiveness of an intervention are discussed, and it was concluded that clinical trials are the best instrument to achieve this goal. 3 In addition to proof of quality, safety, and efficacy of the medicine, each process in a clinical trial ought to be planned, followed through, and automated as much as possible, in order to reduce the risk for the patient and the company to a minimum. In Serbia, as well as around the world, clinical trials represent a very important part of the new medicines’ development process. It has been observed that the number of trials per year has increased significantly over the past 20 years.
Risk Management
The current level of development of both scientific and practical thought on management and pharmacy is the result of the work of a large number of thinkers and practitioners. 4 By combining these two sciences, a special discipline called health management was developed, which can be presented as planning, organization, management, and control, with appropriate human resources policy, in the healthcare system, which is of particular importance for the organization and conduct of clinical trials. 5 In recent years, highly dynamic and intricate social and technological processes have been unfolding, including globalization as a worldwide phenomenon, which has complex causes and far-reaching consequences across various fields, especially in the pharmaceutical industry and, within its scope, in the segment of risk management in clinical trials. Well-founded are the positions of some authors, who suggest that choosing strategies that yield lasting competitive advantage is one of the most significant issues in the effective implementation of strategic management. 6 However, this is by no means a straightforward matter under present conditions with an uncertain future. Accordingly, certain authors point out that with the more recent advancements in technology significant changes have emerged, such as globalization, which has brought forth new techniques and methods in the field of management. Nowadays, management is discussed more as a theoretical and scientific discipline that assists in effectively achieving goals, and somewhat less as a practical skill. 7
Among these theories, the following ones stand out: goal-oriented management, strategic management, system theory, contingency theories, and others. Each of these theories can significantly contribute to clinical trial management efficiency. When considering a clinical trial as a whole composed of multiple segments, it can be useful to mention the system theory, which states that a system consists of interconnected parts that affect one another. 8 Furthermore, the literature suggests that strategic management is particularly useful for organizations operating in dynamic environments, an aspect which any contract research organization (CRO) or pharmaceutical company certainly faces, and that it is crucial to heed faint signals, as it is too late to apply management techniques and methods when the consequences are felt in the operation of the organization. 9 As a distinct tool in strategic management, strategic planning is also worth mentioning, as it represents a subcomponent of the rational approach taken by an organization’s management to achieve strategic objectives. 10 Various factors affect the outcomes of clinical trials, given the complexity of these processes. Due to this complexity, there is a multitude of risks with each step, which can impact not only the company but also the patient. In this regard, establishing a quality management system for clinical trials is vital. This concept can be defined as a set of interconnected subcomponents aimed at efficient and effective organization management. 11 The quality management system also represents an organization’s concept in terms of determining the direction for quality-related activities. 12 With this in mind, for example, two types of risks are observed in quality management, namely, (a) those that can affect the credibility of the clinical trial results and (b) those that can impact the level of safety of individuals subjected to research. 13
Some authors are right to state that legal regulations and bylaws have improved due to higher expectations regarding risk management, but also that they make better assessments due to ever-increasing demands for finding risk-reduction strategies. 14 The above positions are also supported by the studies of the Food and Drug Administration (FDA) of the USA regarding risk management, where the failure mode and effects analysis (FMEA) method plays a significant role. 15
The scientific contribution of this article lies in the fact that there are not many scientific papers on the topic of prospective risk management in clinical trials, and no research on this subject has been conducted in the Republic of Serbia. The goal of this article is to conduct a prospective, qualitative study on risk management in clinical trials in the Republic of Serbia. The study aims to identify, assess, and mitigate risks for patients and companies and to propose corrective measures for the reduction of their frequency and consequences.
Materials and Methods
FMEA
The FMEA method implies “close examination of high-risk processes to identify needed improvements that will reduce the chance of unintended adverse events,” 16 and it can also be defined as a tool that measures risks in all stages of this complex process. 17
The FMEA method was developed by a group of engineers and was initially applied in high-risk industries, such as nuclear power. Recently, the FMEA method has been increasingly applied in the field of healthcare. 18 Accordingly, it is important to point out that the essence of the FMEA method is predicting errors and finding ways to prevent errors. 11
It is important to note that the FMEA method, while valuable, relies on expert judgment and may carry inherent subjectivity in risk scoring.
Study Design
The research was conducted from December 12, 2022 to March 8, 2023. In the course of three months, employee risk data from the CRO were obtained, and then the team met two times in person. First time to analyze the risks and assess them based on severity, probability, and detectability, and second time to review the risks after the theoretical implementation of the proposed corrective measures. The procedure comprised four steps: process selection, team formation, implementation of risk analysis, corrective measure proposal, and risk reanalysis. 18
Steps of the FMEA Method
Process Selection
There are numerous sub-processes within a clinical trial process, most of which are interconnected. The organizer of the study decided not to focus on one specific process or a sub-process within the study, but to attempt to observe the study as a whole instead and to derive conclusions from this entirety, on the processes and sub-processes that should be given attention.
Team Formation
An email was composed and subsequently sent to 10 representative addresses, with a description of the study, the methodology, and a proposal for collaboration. Nine experts from the CRO responded and accepted the cooperation and provided potential risks, with seven experts participating until the end of the research. The expert team consisted of nine individuals, eight of whom were part of the focus group: seven experts from the CRO and one regulatory expert. The organizer of the study was an FMEA method quality and implementation expert and the facilitator of the focus group. Notes were taken by a trained student assistant from the Faculty of Pharmacy at the University of Belgrade, ensuring accurate documentation of the discussions.
Risk Analysis Implementation
Risk analysis implementation consisted of four processes:
Risk identification
Risk identification was conducted by initially having the nine experts identify and deliver the risk data. Following additional analysis of these risks, as many of them overlapped or were without significance in the clinical trials process in Serbia, the number of risks was reduced.
Risk assessment
The assessment was carried out by the expert team, which defined parameters for severity, probability, and detectability for each risk
19
: Severity (S) was ranked on a scale of 1–10, where 1 represented minimal, and 10 represented catastrophic severity; Probability (P) was ranked on a scale of 1–10, where 1 represented minimal probability of risk occurrence, and 10 represented very high probability of risk occurrence; and Detectability (D) was ranked on a scale of 1–10, where 1 represented a very high and 10 represented a very low chance of detecting a risk.
Tables 1–3 describe each segment of the range in more detail.
Severity.

Probability.
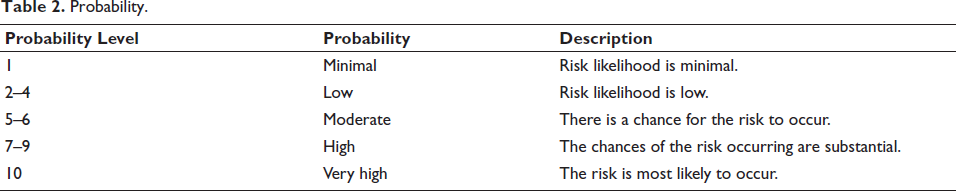
Detectability.
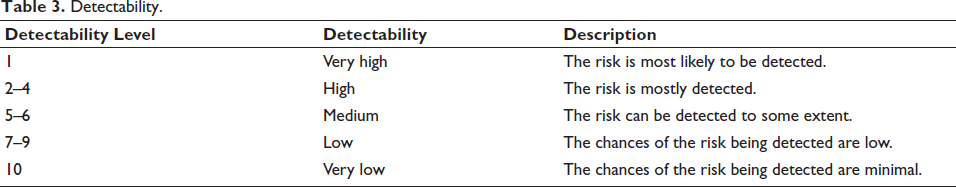
The severity, probability and detectability rating scales (Tables 1-3) were defined according to the general principles of FMEA, as outlined in ICH Q9 and IEC 60812.20 Determining cause and effect
For all high-priority risks, the causes of their occurrence have been defined, along with their effects on other processes and on the overall clinical trial. Calculating risk priority number (RPN)
In the end, the number of risk priorities was calculated for each risk by multiplying the mutual parameters within the same risk. 19 Considering that the grade range for all three parameters is 1–10, the range of RPN value is between 1 and 1,000 (1 × 1 × 1–10 × 10 × 10). Based on the RPN value, the risks were ranked according to priority, from the highest to the lowest priority. Cutoff value, which separates the risks of high, medium, and low priority, was defined. Corrective measures were proposed for the risks whose cutoff value was >100.
Proposed Corrective Measures and Risks Reanalysis
Following the risk analysis that ended with the calculation of the RPN, corrective measures were proposed along with a reassessment of the risks (parameters), along with a recalculation of the RPN for each high-priority risk. Subsequently, old and new RPN values for the same risks were compared.
Results
Initially, 9 experts identified 57 risks, after which the number was reduced to 20 risks, grouped into three categories (conducting trials at research centers, regulatory framework for conducting trials, and negotiating trial conduct with the research centers). The nine-member team proceeded with further analysis.
After the risks were identified, RPN values were obtained, and they varied within the 10–192 range. All 20 risks are ranked in Table 4—research results, from the most to the least significant risks. Considering that the cutoff value was set to be 100, the 5 most significant risks were identified. The risk with the highest RPN value was the inadequate process of obtaining informed consent from research subjects, including the inability to implement current solutions (e.g., eConsent; RPN = 192). All of the listed risks are related to the trial conduct at research centers. The remaining four are as follows: Insufficient transparency/availability/interconnection of subjects’ medical data (RPN = 126), Insufficient equipment and inadequate capacity of a significant number of research centers.
Research Results.
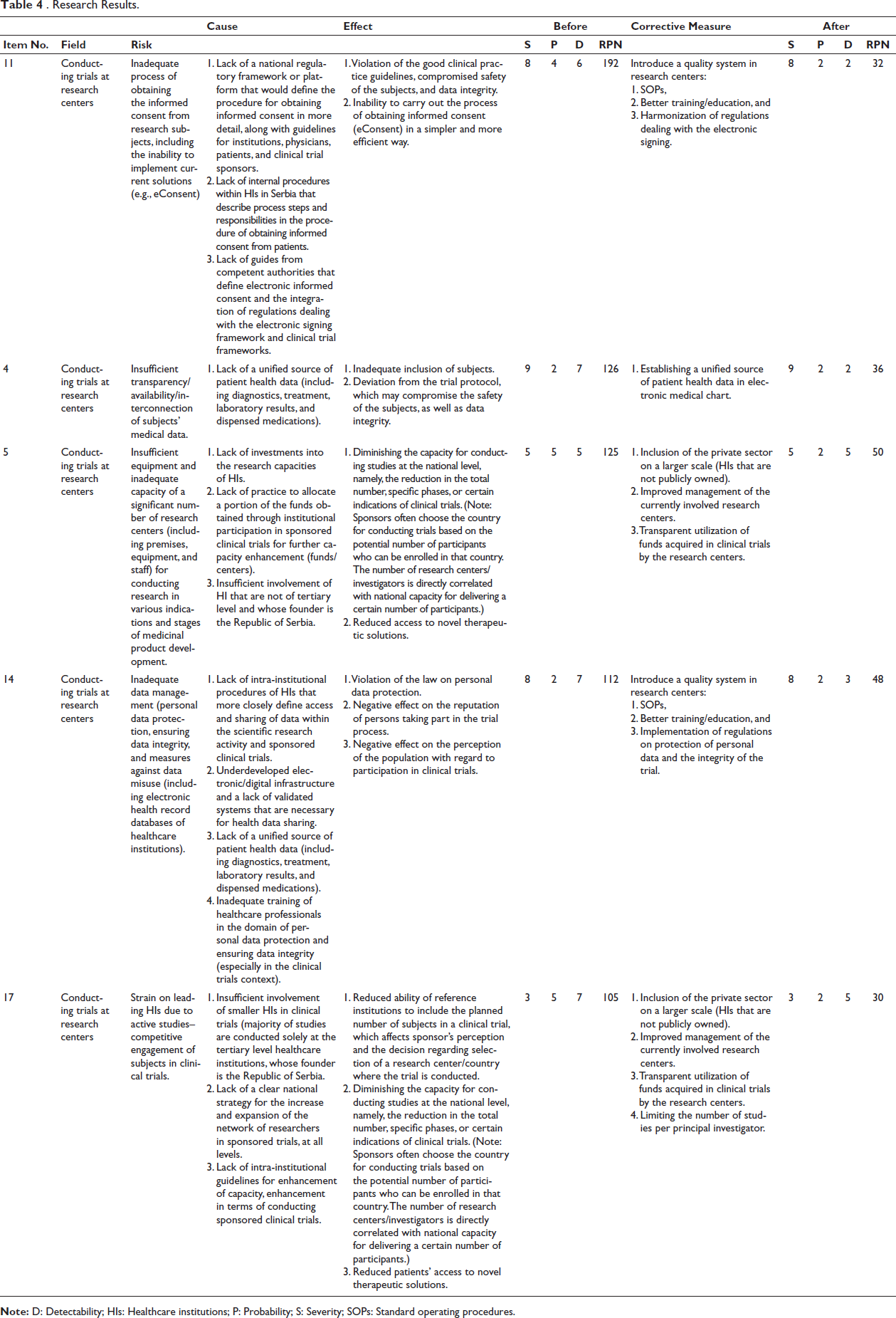
(Including premises, equipment, and staff for conducting research in various indications and stages of medicinal product development (RPN = 125)), Inadequate data management (personal data protection, ensuring data integrity, and measures against data misuse, including electronic health record databases of Healthcare Institutions (HIs); RPN = 112), and Strain on leading HIs due to active studies–competitive engagement of subjects in clinical trials (RPN = 105).
Given that every clinical trial sub-process affects other processes and the whole, it is not simple to clearly separate the causes and consequences of risks. Certainly, for research purposes, 12 causes and 19 effects were then identified for the aforementioned risks.
Causes
It can be observed from the table that within the framework of the 12 causes, two have an impact on multiple risks. These are: Insufficient involvement of HIs that are not of tertiary level and whose founder is the Republic of Serbia and Lack of a unified source of patient health data (including diagnostics, treatment, laboratory results, and dispensed medications).
For the risk with the highest RPN value (inadequate process of obtaining the informed consents from research subjects, including the inability to implement current solutions (e.g., eConsent)), three causes have been identified: Lack of a national regulatory framework or platform that would define the procedure for obtaining informed consent in more detail, along with guidelines for institutions, physicians, patients, and clinical trial sponsors, Lack of internal procedures within HIs in Serbia that describe process steps and responsibilities in the procedure of obtaining informed consent from patients, and Lack of guides from competent authorities that define electronic informed consent and the integration of regulations dealing with the electronic signing framework and clinical trial frameworks.
Effects
Various situations can occur in a single clinical trial. Some of the consequences of the most significant risks are as follows: Violation of the good clinical practice guidelines, compromised safety of the subjects, and data integrity, Inability to carry out the process of obtaining informed consent (eConsent) in a simpler and more efficient way, Inadequate inclusion of subjects, and Deviation from the trial protocol, which may compromise the safety of the subjects, as well as data integrity.
All of the above effects, as well as the rest of the effects in Table 4—research results, significantly affect not only the quality of the trial but also the safety of the subjects.
Corrective Measures
Corrective measures within the FMEA method are suggestions for minimizing the severity and occurrence and maximizing the detectability of a given risk. A total of seven corrective measures were proposed for the five most significant risks. Some of the measures that can be implemented for multiple risks are as follows: Introduce a quality system in research centers (standard operating procedures; SOPs, better training/education), Include the private sector on a larger scale (HIs that are not publicly owned), Improve management of the currently involved research centers, and Transparently utilize funds obtained by research centers in clinical trials.
After proposing corrective measures, the team reassessed the risks and calculated RPN values for each of the five most significant risks. The minimum reduction in RPN risk value was 57.14%, and the maximum was 83.33% when compared to the initial values. Also, the total reduction of the RPN value of the five most significant risks was 70.6%.
The maximum reduction was achieved with the inadequate process of obtaining informed consent from the subjects, including the impossibility of implementing current solutions (e.g., eConsent), namely, by implementing a quality system in research centers.
Corrective measures were designed to directly address the identified risk categories—trial conduct at research centers, regulatory framework, and negotiation processes—thereby enhancing both the clarity and the practical applicability of the proposed solutions.
Discussion
In the conducted trial, numerous potential risks that can emerge during a clinical trial were identified, and corrective measures were proposed for the most significant risks (risks with the highest RPN values). The study revealed that the FMEA method can substantially reduce the probability of risk occurrence and increase the chance of its detection, resulting in a reduction of the RPN.
It has been demonstrated that the most significant risks are primarily in the field of conducting trials at research centers, and within this field, risks associated with information acquisition methods, their management, and the equipment level of research centers were emphasized. As the most significant risks manifest at the outset of the clinical trial process, they can exert a substantial impact on the entire trial. It can also be concluded that by addressing potential risks more attentively at the beginning of the trial and by implementing corrective measures (such as introducing quality systems in research centers and improving research center management), the likelihood of encountering such risks can be significantly reduced, and their detectability increased.
Some of the advantages of this method have been shown to be the following: Approaching problems from multiple angles. Prospective analysis. Tri-faceted analysis (severity, probability, and detectability). Corrective measures that can be evaluated both in theory and practice.
Some of the drawbacks of the above method are related to the subjectivity inherent in the assessment process. Hence, comparing the RPN with the results obtained from other analyses is challenging. Some authors deem that if processes are similar, trends identified in different trials are comparable. 17 Due to this subjectivity, a greater number of such studies are required to assess risks with greater precision. Also, the study was not conducted in a clinic. It was based on a theoretical overview of the identifying, assessing, and reducing risks based on the knowledge and previous experience of experts.
Nevertheless, one of the advantages of this study is deemed to be the fact that not only clinical trial experts are involved, but also experts in quality control and regulatory experts, with technical support from a student who had no previous practical experience in the given fields.
Considering that there are not enough papers in the world that analyze prospective management in the clinical trial process, there was not sufficient literature to which the results could be compared. Nevertheless, the paper, FMEA drastically reduced potential risks in clinical trials conducted by Lee et al., can be used as a good starting point for discussion. 13
Table 5 presents a comparison of the results obtained from this study with the research conducted by Lee et al.
13
In our article, risks with the highest RPN values were attributed to a single field, while Lee et al. identified three fields (staff training at the site, document management, including source documents and main documents, and obtaining informed consent), and the two most significant are as follows: Using outdated informed consent forms, RPN = 378 and Failing to update training records, RPN = 315.
Comparison of Scientific Papers.
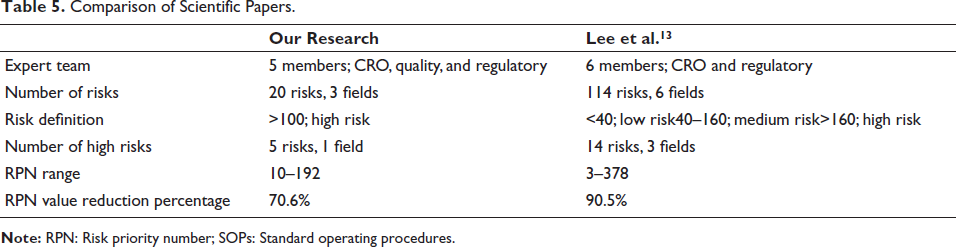
In both articles, changes in probability and detectability accounted for differences in RPN values following the implementation of corrective measures and re-evaluation of risks. On the other hand, severity did not change significantly.
Limitations
This study has certain limitations. First, it was theoretically designed and not validated through application in an actual clinical trial setting. Second, the expert group was relatively small, which may limit the generalizability of the findings. Third, the FMEA method inherently involves subjective judgment in risk scoring, despite efforts to ensure consensus among experts. These limitations suggest that the results should be interpreted with caution and verified in future applied studies.
Conclusion
Due to their complexity, the processes in clinical trials require not only mutual alignment but also strategic and systematic management, demanding a multidisciplinary and synergistic approach across various scientific fields. With the increasing number of trials and growing experience, these processes are becoming regulated in more detail, both at the state and company levels, which somewhat facilitates their management. However, the FMEA method has demonstrated that certain risks can still be better controlled. It is believed that the potential implementation of corrective measures would greatly reduce each of the significant risks.
Based on previous studies, the process of obtaining informed consent is the most significant risk, where corrective measures can affect the probability and detectability of risk occurrences. The proposed measures can serve as a foundation for developing risk management protocols in Serbian CROs and HIs. Future studies should consider applying the proposed measures in a clinical setting to validate their effectiveness in real-world conditions. Considering that there are not enough papers that analyze prospective risk management in the process of clinical trials, this study could represent a step forward and a foundation for future research.
Footnotes
Abbreviations
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.