
Abstract
Background:
Parkinson’s disease (PD) is a movement-affecting neurodegenerative condition with an unclear etiology. Recent research suggests targeting poly-(adenosine 5-diphosphate-ribose) polymerase 1 (PARP1) as a potential therapeutic approach for PD treatment.
Purpose:
The purpose of this study is to assess the effect of an ethanolic extract of Moringa oleifera leaves (MOE) on a 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced Parkinsonian mouse model, with a specific focus on investigating its potential to mitigate the effects of α-synuclein toxicity, oxidative stress–induced hyper-activation of PARP1, and mitochondrial dysfunction associated with PD pathology. Additionally, this study also intends to investigate the alterations in neurobehavioral and biochemical parameters associated with PD pathology.
Materials and Methods:
An in silico docking study was conducted to investigate the phytochemicals found in M. oleifera (MO, drumstick plant) leaves as the potent inhibitors of PARP1. An in vivo (neurobehavioral, biochemical, and western blot) study was conducted to assess the neuroprotective effect of MOE on the MPTP-induced Parkinsonian mouse model.
Results:
The results of in silico study showed that the phytochemicals found in MO leaves could be a potent inhibitor of PAPR1. The in vivo study results showed that MOE significantly ameliorated MPTP-induced neurobehavioral and biochemical deficits. MPTP-induced mitochondrial enzyme-complex deficits were found to be restored in MOE-treated mice. Additionally, the result obtained in the western blot analysis showed that MOE significantly restored the levels of tyrosine hydroxylase in MPTP-intoxicated mice. MOE enhanced the expression of the anti-apoptotic factor (Bcl-2) and suppressed the expression of pro-apoptotic factors (Bax and caspase-3). Additionally, the enhanced levels of α-synuclein and PARP1 were significantly suppressed by MOE.
Conclusion:
Our findings suggest that MOE may possess pharmacological properties that inhibit neuronal damage in MPTP-intoxicated mice. Thus, MOE could be used as a therapeutic agent that can protect dopaminergic neurons from PARP1-induced neuronal damage.
Introduction
The selective and progressive loss of dopaminergic neurons and the aggregation of the misfolded protein α-synuclein in the substantia nigra lead to the second most common neurodegenerative disorder, known as Parkinson’s disease (PD). PD is characterized by various motor abnormalities such as balance impairment, resting tremor, bradykinesia, and rigidity. 1 The currently available treatment regimen alleviates only the motor symptoms but plays no role in arresting neuronal loss. Also, the drug’s efficacy is observed to be diminishing over time, and various major side effects have also been reported. Although the precise etiology of the disease is still unknown to date, most of the experimental studies suggest that oxidative stress and mitochondrial dysfunction play a crucial role in disease progression. 2
In the recent past, Kam et al. have defined a new mechanism driven by poly-(adenosine 5-diphosphate-ribose) (PAR) and poly-(adenosine 5-diphosphate-ribose) polymerase (PARP1) that leads to α-synuclein toxicity. 3 A selective environmental neurotoxin, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), has been widely used to induce Parkinson’s-like motor dysfunction by inducing dopaminergic cell loss in substantia nigra pars compacta (SNpc). MPTP leads to the aggregation of intra-neuronal eosinophilic inclusions, markers of escalated oxidative stress, and a decrease in mitochondrial complex 1 activity, just like PD. Nitric oxide and superoxide anions have been believed to play a major role in MPTP neurotoxicity via an unknown mechanism. Peroxynitrite-driven O−2- and NO-mediated neurotoxicity leads to cell death via both apoptotic and non-apoptotic morphologies. 4 Excessive peroxynitrite, nitric oxide, and superoxide anions lead to DNA damage and are the only common downstream pathways leading to neuronal loss. Extensive DNA damage over-activates PARP1, which under normal conditions engages itself in DNA repair by transferring the adenosine triphosphate (ADP) ribose group to several nuclear proteins, utilizing nicotinamide adenine dinucleotide (NAD) as a substrate. 5 PARP1 frequently causes cell death by depleting the cellular energy store by depreciating the storage of cellular NAD and ATP’” with “PARP1 frequently causes cell death by depreciating the storage of cellular NAD and ATP. PARP1 has also been shown to act directly on the mitochondria, triggering peroxynitrite-induced mitochondrial damage. As a result, it was postulated that suppressing the activity of over-activated wild-type PARP1 could treat neurodegenerative disorders. 6
PARP1 inhibitors have been shown to be effective in a PD model; however, the function of PARP1 in the pathophysiology of PD is as yet unknown. MPTP primarily activates PARP1 in susceptible dopaminergic neurons, and PARP1 gene knockout mice are significantly protected from MPTP neurotoxicity. 7 These findings imply that PARP1 inhibitors may be effective in the treatment of PD. The therapeutic efficacy of PARP1 inhibitors has been studied in various neurodegenerative disease models including Alzheimer’s 8 and Parkinson’s diseases. 9
This study focusses on the identification of potential inhibitors of PARP1 from phytochemicals present in Moringa oleifera (MO, drumstick plant) leaves, a traditional medicinal herb known for its nutritional, medicinal, and neuroprotective properties, 10 via an in silico docking study analysis. The inhibition potency of the phytochemicals has been compared with the inhibition potency of known PARP1 inhibitors Talazoparib (BMN 673), PJ34, and FR257517. An in vivo study was conducted to understand the therapeutic effect of MO leaf extract (70% ethanol) on the neurobehavioral and biochemical parameters. The effect of MO leaf extract on mitochondrial functions was also studied. A western blot analysis was conducted to measure the expression of tyrosine hydroxylase (TH), PARP1, caspase-3, and α-synuclein.
Materials and Methods
In silico Docking Study
Selection and Preparation of Inhibitors/Ligands and Receptor Protein (PARP1)
The phytochemicals used in this study were selected based on the available literature. These phytochemicals were retrieved from the NCBI PubChem database. The crystal structure of the constitutively active protein PARP1 (Protein Data Bank (PDB) ID: 5A00) was retrieved from Research Collaboratory for Structural Bioinformatics (RCSB)-PDB in the .pdb format. The ligands were downloaded from PubChem in the .sdf format and were converted to .pdb and .pdbqt formats using Open Babel and AutoDock tools, respectively. For docking studies, the protein was prepared using AutoDock tools. The water molecules were deleted; polar hydrogen was added along with the addition of Kollman charges, fragmental volumes, and solvation parameters; and the file was saved in the .pdbqt format.
Molecular Docking Studies
The active binding site was predicted using the Computed Atlas of Surface Topography of proteins (CASTp), and the grid box was prepared using AutoGrid. The grid size was designated as 40 × 40 × 40 xyz points with 0.347 grid spacing, and the dimensions (x, y, and z) of the grid center were set to be 20.918, 21.868, and 71.882. AutoDock Vina was used for docking. An iterated local search global optimizer was utilized by AutoDock Vina, and in the complete docking procedure, both the ligands and protein were considered rigid. The two-dimensional image of the docked structure (protein–ligand interaction) was generated using LigPlot+.
In vivo Experimental Studies
Reagents and Antibodies
Bovine serum albumin (BSA), disodium hydrogen phosphate, potassium chloride, ammonium chloride, digitonin, reduced nicotinamide adenine dinucleotide phosphate (NADPH), mannitol, and oxidized cytochrome c were procured from Sisco Research Laboratories (SRL, Mumbai, India). MPTP and 2-nitrobenzoic acid (DTNB) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Primary antibodies for PARP1 were purchased from Elabscience (TX, USA), and primary antibodies for TH, β-actin, and Bax were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Primary antibodies for Bcl-2 were procured from Abcam Life Science, Biogenuix Medsystems Pvt. Ltd. (New Delhi, India).
Plant Material Collection and Preparation of the Plant Extract
Fresh leaves of MO were collected from the campus of BHU, Varanasi (India), during the months of March and April 2020. The fresh leaves were washed, cleaned, and cut into small pieces before being shade-dried. Subsequently, the dried leaves were ground into a fine powder. This powder was then subjected to extraction using 70% ethanol in a flat-bottom flask at room temperature, and the mixture was left to stand for several days until the solvent became concentrated. The concentrated solvent was filtered through a Whatman filter paper, and the resulting filtrate was further concentrated using a rotary evaporator at 40°C. The concentrated extract was subsequently dried and stored at 4°C. Before being used, the semi-solid extract was dissolved in distilled water.
Experimental Animals
Male Swiss albino mice (25 ± 5 g) used for the experimental study were procured from the animal facility at the Institute of Medical Sciences, BHU, Varanasi (India). Mice were kept in clean polypropylene cages and were allowed to acclimatize to the animal facility for a week. They were provided with a constant supply of standard diet pellet and water ad libitum.
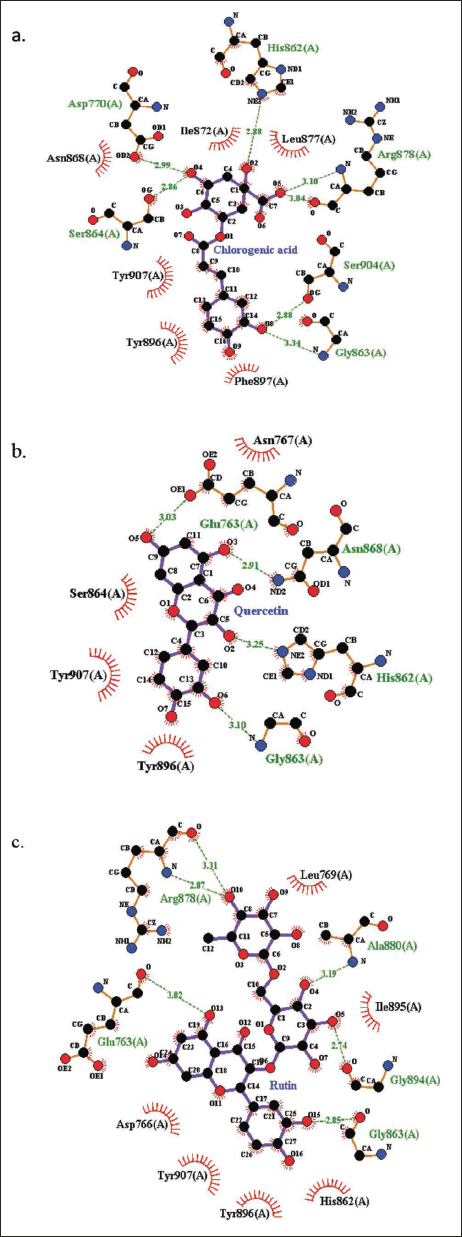
Animal Dosing
The experimental animals were divided randomly into four groups with 6 mice/group: control, MPTP, MPTP + MO extract (treatment group), and MO extract (positive control). Group 1: Normal control—for 24 days, the mice were given 0.9% normal saline orally once a day; Group 2: Disease control (DC) group—given doses of MPTP (30 mg/kg body weight), dissolved in normal saline for 5 consecutive days via intraperitoneal injection (day 20 to 24 of ethanolic extract of M. oleifera leaves (MOE) dosing); Group 3: Treatment group—the treatment doses of MOE 400 mg/kg body weight was administered daily for 24 days via the oral route, and MPTP was administered intraperitoneally in a similar manner as done for Group 2; Group 4: Positive control—animals received oral administration of 400 mg/kg MOE for 24 consecutive days (once daily). After the dose completion, mice were subjected to a neurobehavioral test from day 25 to 28. After the behavioral analysis, mice were sacrificed, and the brains were isolated for biochemical analysis, mitochondrial dysfunction analysis, and western blot analysis.
Neurobehavioral Analysis
After the completion of the dosing cycle, mice were subjected to training for various neurobehavioral analyses in order to evaluate the motor function impairment induced due to MPTP intoxication. Four behavioral tests including rota-rod, traction, pole, and catalepsy bar test were conducted.
Rotarod Test.
Mice from all the groups were trained for 3 consecutive days on a rotarod (speed: 5 rpm). The time spent by each mouse on the rotarod was recorded. The training was given 4 times a day, and the average time spent by the mice was recorded. 11 After the completion of the dosing and the training period, mice were again taken for the rotarod test, and the time spent by each mouse on the rotarod was recorded individually.
Traction Test.
The ability of the mice to hold a horizontally hanging bar using their front paws is measured in the traction test. The bar is usually placed 40 to 50 cm above the tabletop so that the animal has enough time and space when falling to land on its feet with the righting reflex. 12 The scoring was done from 1 to 3 on the basis of the hind limb placement.
Pole Test.
In this test, mice are placed on the top of a pole, and the time taken by the mice to orient downward and descend is measured. 12 After the completion of the final dosing of MPTP, mice were subjected to the pole test by placing them on the top of the pole with a rough surface. As soon as the animal began to turn, the recording was started. Both the T-turn (time to turn completely downward) and T-total (time to reach the floor) were recorded accordingly.
Catalepsy Bar Test.
The mice were made to stand in an unusual posture by placing their forelimb on the wooden bar (height 5 cm from the ground) and the hind limbs on the ground. 13 The time taken by the mice to correct their unusual posture was recorded. The maximum time given to correct the unusual posture was 3 minutes.
Assessment of Mitochondrial Parameters
Isolation of Mitochondria.
Brain mitochondrial isolation was carried out via differential centrifugation. 14 Mice were sacrificed by cervical dislocation followed by decapitation, and the mid-brain was isolated. The mid-brain of the mice was washed thoroughly and homogenized in a homogenizing buffer (mM HEPES, 1 mM ethylene glycol tetra acetic acid (EGTA), 225 mM mannitol, 1 mg/ml BSA (pH 7.4), and 75 mM sucrose). The homogenates were centrifuged at 2,000g at 4oC for 30 minutes. The supernatant was collected and centrifuged at 12,000g at 4oC for 10 minutes. The pellet was collected and suspended in a homogenizing buffer containing 0.02% digitonin to separate the mitochondria from synaptosomal fractions. The suspended mixture was again centrifuged at 12,000g at 4oC for 10 minutes. The pellet was collected, which consists of crude mitochondria, and washed twice with a homogenizing buffer lacking BSA and EGTA. The pellet was finally re-suspended in 50 mM phosphate buffer, and the protein was estimated by the Bradford assay. All assays were conducted within 24 hours of mitochondrial isolation using 20 μg proteins.
Complex I (NADH Dehydrogenase) Activity Assay.
The enzymatic activity of NADH dehydrogenase was estimated as per the following standard protocol. 15 In brief, the reaction mixture was prepared by adding 20 µg of mitochondrial protein in 100 µl of potassium phosphate buffer (0.5 M, pH 7.5), 30 µl of KCN (10 mM), 60 µl of fatty acid–free BSA (50 mg ml−1), and NADH 10 µl (10 mM). The content was mixed properly, and the absorbance was taken at 340 nm for 2 minutes. The extinction coefficient of NADH at 340 nm is 6.2 mM−1 cm−1. The enzyme activity was expressed in nmol min−1 mg−1.
Complex II (Succinate Dehydrogenase) Activity Assay.
The enzymatic activity of succinate dehydrogenase (SDH) was estimated by using the following standard protocol 15 with few changes. Briefly, the reaction mixture consisted of 50 µl of 0.5 M potassium phosphate buffer, 50 µl of 400 mM succinate, 30 µl of 10 mM potassium ferricyanide, 20 µl of fatty acid–free BSA, and nitroblue tetrazolium (NBT). 20 µg of mitochondrial protein was added to the reaction mixture, and the reaction was initiated. After mixing the contents properly, absorbance was taken at 600 nm for 2 minutes, and the enzymatic activity was expressed as µM of formazan produced min−1 mg−1 protein.
Complex IV (Cytochrome C Oxidase) Activity Assay.
Before the determination of the complex IV activity, the oxidized cytochrome c solution was first converted to reduced cytochrome c using sodium dithionite. The reaction mixture was prepared by adding 20 µg mitochondrial protein to a solution of 0.5 M potassium phosphate buffer and 0.3 mM reduced cytochrome c. The enzymatic activity was determined by the decline in absorbance at 550 nm for 3 minutes. The result of cytochrome c oxidase activity was expressed in nmol min−1 mg−1 protein. The extinction coefficient at 550 nm was taken as 19.6 mmol−1 cm−1. 15
Complex V (F1F0 ATP Synthase) Activity Assay.
The F1F0 ATP synthase activity was determined as per the standard protocol. 16 In brief, the reaction mixture consisted of the mitochondrial protein sample and ATPase buffer (5 mM ATP, 2 mM MgCl2, and Tris-HCl, pH 8.5). The reaction mixture (1 ml) was incubated at 30oC for 5 minutes, and then 10% trichloro acetic acid (TCA) was added to it, followed by centrifugation for 10 minutes at 3,000g. As a result of ATP being hydrolyzed into ADP, inorganic phosphorus (Pi) was released. The liberated Pi was measured in this assay and was expressed in nmol of Pi liberated min−1 mg−1 protein.
Mitochondrial Manganese Superoxide Dismutase Assay.
The estimation of manganese superoxide dismutase (Mn-SOD) was done as per the prescribed protocol. 17 The reaction mixture was prepared by adding 0.1 mM EDTA, Triton-X (0.6%), 50 mM sodium carbonate, 90 mM NBT, and hydroxylamine hydrochloride to the suspended mitochondrial sample. Superoxide was produced as a result of photooxidation of hydroxylamine hydrochloride. Superoxide ions underwent further reduction of nitroblue tetrazolium (NBT), and this reaction was inhibited by the presence of superoxide dismutase (SOD). The absorbance was recorded at 560 nm for 3 minutes, and the activity of SOD was expressed in IU mg−1 protein (1 IU = quantity of enzyme necessary for 50% inhibition).
Mitochondrial GSH (mt-GSH) Assay.
The reduced GSH content in the mitochondria was estimated as per the prescribed protocol. 17 In brief, 20% of trichloroacetic acid was added to 20 µg of the mitochondrial sample and was centrifuged at 1,500g. The pellet was discarded, and 5,5-dithiobis 2-nitrobenzoic acid (DTNB) was added to the supernatant. DTNB reacts to the protein’s thiol group and forms 2-nitro-5-mercapto benzoic acid. The absorbance of the reaction mixture was taken at 412 nm. A standard curve was prepared by commercially accessible GSH, and the content was expressed in mmol g−1 of mitochondrial protein.
Western Blot Analysis
After the completion of the dosing, mice were sacrificed and their brains were isolated. The substantia nigra of the brains was homogenized in a RIPA (lysis) buffer, and the homogenate was subjected to centrifugation (12,000 rpm for 30 minutes). The supernatant was collected, and the protein was quantified using Bradford assay. A total of 40 µg of protein was loaded on to each well of 10% polyacrylamide gel, and after the completion of electrophoresis, proteins were transferred to a PVDF membrane. The membrane was left overnight for incubation in 1o antibodies TH (1:1,000), Bax (1:1,000), Bcl-2 (1:800), caspase-3 (1:1,000), PARP1 (1:1,500), α-synuclein (1:1,000), and β-actin (1.500) at 4oC. After overnight incubation, the blots were washed thrice (10 minutes each) in TBST, and the membrane was further incubated in an HRP-conjugated secondary antibody for 2 hours at room temperature. The membrane incubated in the secondary antibody was washed twice in TBST. After washing, the blots were developed using ECL as a substrate (luminol + H2O2) and were visualized in a Biorad gel doc imager (428 nm). Quantity One software (Biorad, Windows) was used to identify the band density, and the relative band density was calculated with respect to that of β-actin.
Statistical Analysis
The statistical analysis of the data was carried forward by one-way analysis of variance (ANOVA) using the Newman–Keuls Multiple Comparison test via the GraphPad Prism 5.0 software. The results were articulated in terms of mean ± SEM. The outcomes were expressed using the mean ± standard error of the mean (SEM), with statistical significance attributed to p values less than 0.05.
Results
In silico Result Analysis
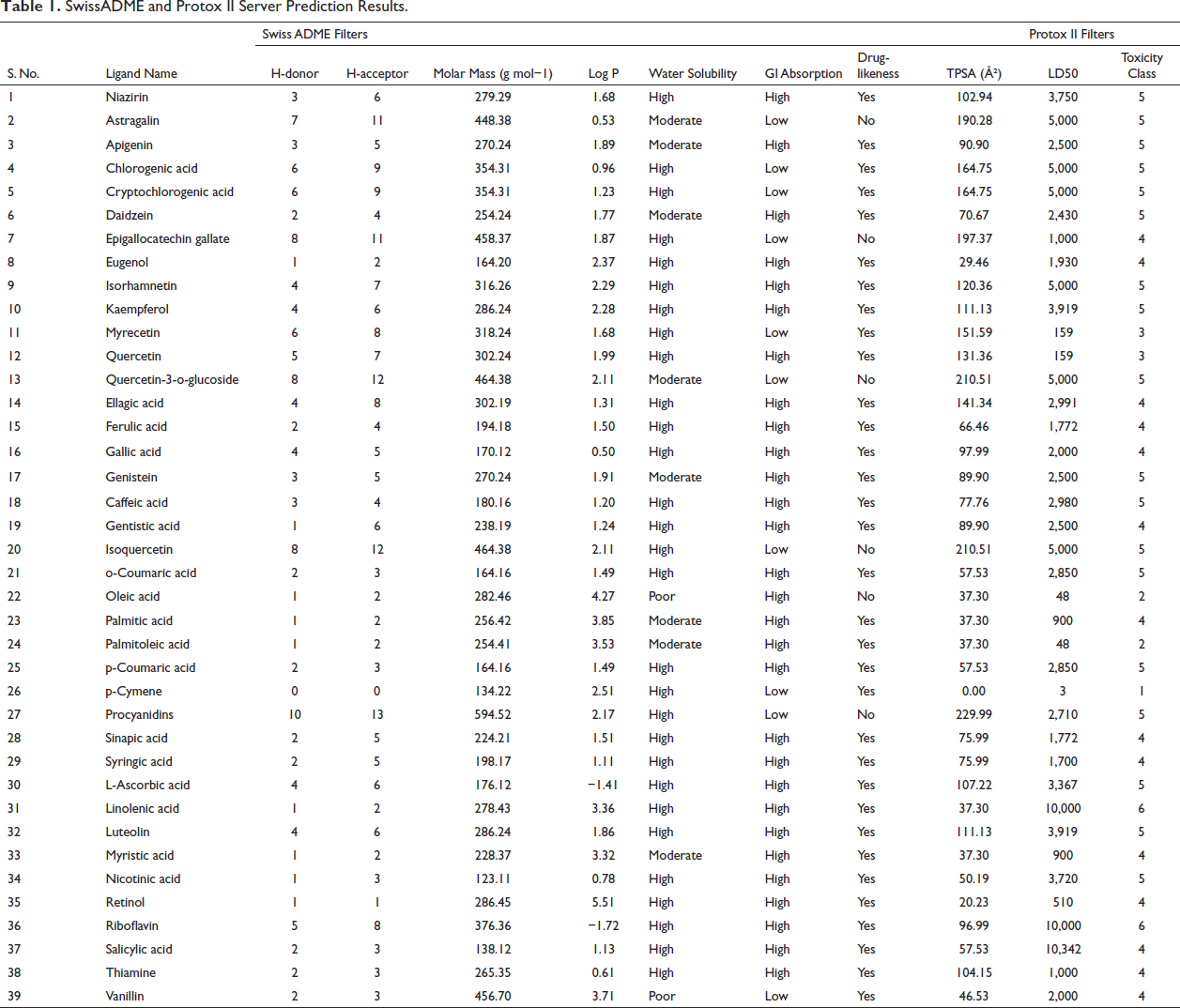
Results of SwissADME and Protox II Server
The phytochemical constituents of MO were retrieved from PubChem, and their SMILE format was submitted to SwissADME and Protox II servers to investigate its drug-like properties. A majority of the phytochemicals were observed to pass both SwissADME and Protox II filters, as shown in Table 1. The compounds were checked for their drug-like properties (the Lipinski rule of five), and it was observed that 33 out of 39 phytochemicals passed this criterion as per the SwissADME prediction. Phytochemicals with a topological polar surface area (TPSA) value less than 140 Å2 are considered to be easily absorbed by the intestine, blood–brain barrier permeant, and bioavailable. The TPSA value of the ligands and the results of Protox II are summarized in Table 1. The TPSA values of 31 out of 39 phytochemicals were found to be less than 140.
SwissADME and Protox II Server Prediction Results.
Binding Energy Analysis (Inhibition Potential) of M. oleifera Phytochemicals with PARP1 Protein
Docking studies were conducted using AutoDock Vina. All those ligands that passed the drug-likeness parameters of SwissADME and Protox II were carried forward for docking against the catalytic site of PARP1 protein and were compared to the docking results of the reported PARP1 inhibitors, that is, PJ34, Talazoparib, and FR257517. The results were analyzed on the basis of various parameters such as binding energy, hydrogen bond formation, and root mean square deviation (RMSD value), as summarized in Table 2. Among the 33 ligands, 19 ligands depicted high binding affinity in comparison to that of reported PARP1 inhibitors. The H-bond interaction of the best three protein–ligand interactions was generated using LigPlot+ (Figure 1).
Result of the Docking Study of PARP1 and Ligands.
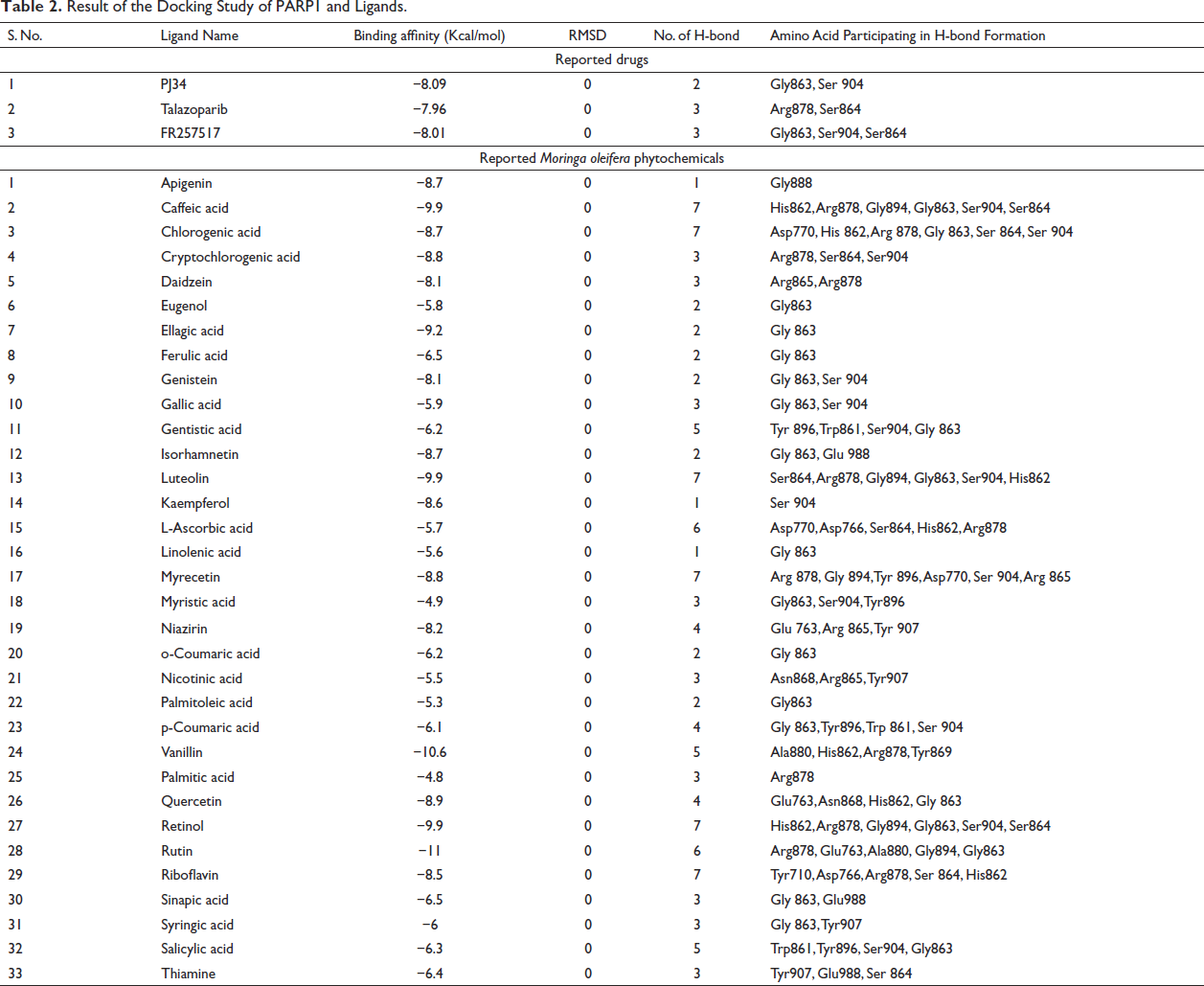
H-bond Interaction by LigPlot+ (a) Between PARP1 and Chlorogenic Acid, (b) Between PARP1 and Quercetin, and (c) Between PARP1 and Rutin.
Animal Dosing Pattern.
In vivo Result Analysis
Effect of M. oleifera on MPTP-induced Neurobehavioral Deficits
Mice were subjected to the rotarod test on day 25, after the completion of the dosing cycle. It was found that MPTP-intoxicated mice spent significantly lesser time (p < 0.001) on the rotarod as compared to the control group. MOE pre-treatment reports showed significant increase (p < 0.001) in the retention time in the treatment group (Figure 3(a)).
Neurobehavioral Analysis of Mouse: MOE Ameliorates MPTP-induced Neurobehavioral Deficit. (a) Time Latency on a Rotarod, (b) Traction Score in the Traction Test, (c) T-turn Time in the Pole Test, and (d) Catalepsy Bar Test. The Values Are Expressed in Terms of Mean ± SEM (n = 5). *p < 0.05; **p < 0.01, and ***p < 0.001. The Results Were Analyzed via One-way ANOVA Followed by the Newman–Keuls Test Using the GraphPad Prism Software.
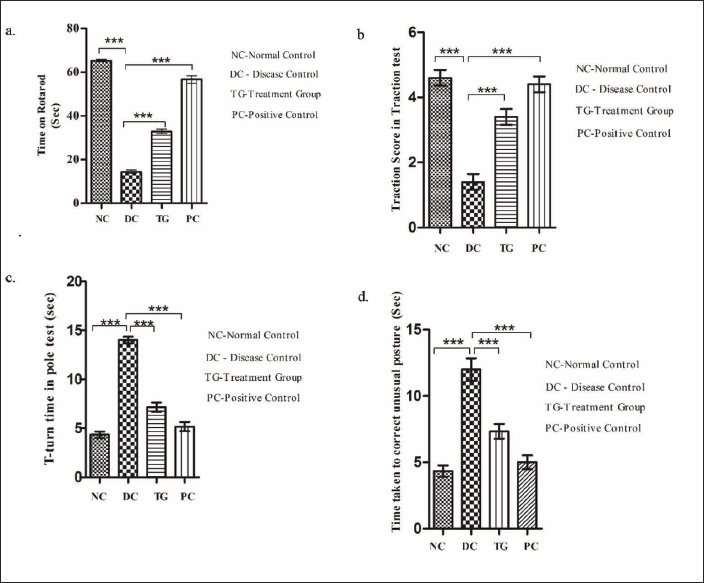
The traction score in MPTP-intoxicated mice was found to be significantly lower (p < 0.001) than that of the control group, signifying reduced neuromuscular strength and weak hind limb grip in the diseased group. The MOE pre-treated group showed a significantly increased traction score and strong hind limb grip (p < 0.001) (Figure 3(b)).
In the pole test, it was found that in MPTP-intoxicated mice, the T-turn time was significantly higher (p < 0.001) as compared to the control group, which signifies prolonged motor dysfunction in the diseased group. In the MOE pre-treated group, the observed T-turn time was significantly reduced (p < 0.001), signifying improvement in MPTP-induced locomotor dysfunction by the MOE treatment (Figure 3(c)).
In the catalepsy bar test, the time taken by the MPTP-intoxicated mice to regain their normal posture was observed to be significantly higher (p < 0.001) as compared to the control group. In the MOE pre-treated group, the time taken to correct their unusual posture was observed to be significantly less (p < 0.001), suggesting improvement in MPTP-induced bradykinesia by MOE treatment (Figure 3(d)).
No significant difference was observed in the positive control group that received oral administration of the MO extract in comparison to the saline-treated normal control group.
Effect of M. oleifera Leaf Extract on Mitochondrial Parameters
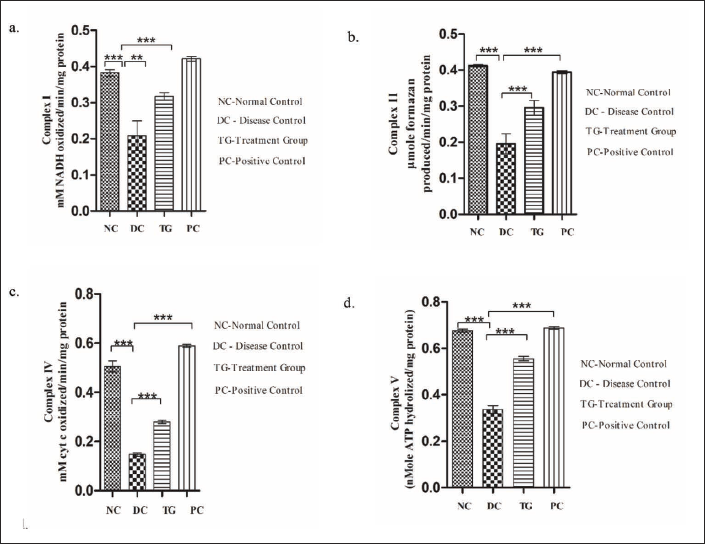
Effect of M. oleifera Leaf Extract on MPTP-induced Changes in Mitochondrial Complex I, II, IV, and V Activity.
Mitochondria is the source of energy in the cell. The enzyme complex (I, II, III, IV, and V) present in the inner mitochondrial membrane undergoes the process of oxidative phosphorylation to produce ATP. Functional loss to any of the enzyme complex can lead to adverse consequences, including loss in ATP. The effect of MPTP on the enzyme complex of the respiratory chain was analyzed as MPTP is known to inhibit complex I of electron transport chain (ETC). The level of mitochondrial complexes (I, II, IV, and V) was observed to vary significantly among different groups as shown in Figure 4. The first enzyme of ETC (NADH dehydrogenase) was found to be significantly decreased (p < 0.001) in the MPTP-intoxicated group as compared to the control group. However, in MOE pre-treated mice, the level of complex I enzyme was observed to be significantly enhanced (p < 0.01) in comparison to the diseased group (Figure 4(a)). A significant reduction was observed in the level of complex II (p < 0.001) (Figure 4b), complex IV (p < 0.001) (Figure 4(c)), and complex V (p < 0.001) (Figure 4(d)) activity as compared to the control group. Similarly, mice that received MOE pre-treatment were observed to have significant enhancement in the level of complex II (p < 0.001), complex IV (p < 0.001), and complex V (p < 0.001) in comparison to the diseased group. No significant difference was observed between the saline-administered control group and MOE-administered positive control group.
Effect of MOE on MPTP-induced Mitochondrial Alterations. (a) Complex I, (b) Complex II, (c) Complex IV, and (d) Complex V. Values Are Expressed in Terms of Mean ± SEM (n = 5). *p < 0.05, **p < 0.01, ***p < 0.001. One-way ANOVA Was Used to Analyze the Result, and Further Analysis Was Performed Using the Newman–Keuls Test.
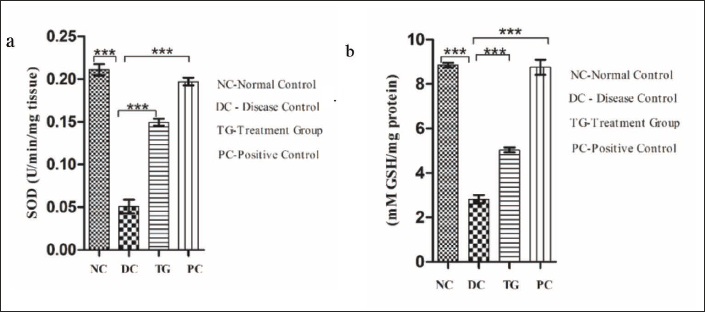
Effect of M. oleifera Leaf Extract on Mitochondrial Superoxide Dismutase and Glutathione.
An increase in the level of oxidative stress (MPTP intoxication) leads to the functional loss of ETC, as a result of which the level of mitochondrial Mn-SOD and GSH (mt-GSH, reduced) declines (Figure 5). The level of Mn-SOD was observed to be significantly decreased (p < 0.001) in the MPTP-intoxicated group as compared to the control group (Figure 5(a)). Similarly, the level of mt-GSH was observed to be significantly reduced (p < 0.001) in the MPTP-intoxicated group in comparison to the normal control group (Figure 5(b)). In contrast, in the MOE-treated group, a significant increase in the level of Mn-SOD (p < 0.001) and mt-GSH (p < 0.001) was observed as a result of the radical scavenging property of MOE.
Effect of MOE on Mitochondrial Mn-SOD and mt-GSH: (a) Mitochondrial Superoxide Dismutase Level; (b) Mitochondrial Reduced Glutathione Level. Values Are Expressed in Terms of Mean ± SEM (n = 5). *p < 0.05, **p < 0.01, ***p < 0.001. One-way ANOVA Was Used to Analyze the Result, and Further Analysis Was Performed Using the Newman–Keuls Test.
Effect of MOE on Tyrosine Hydroxylase, α-Synuclein, PARP1, and Mitochondrial Dysfunction–Induced Apoptosis
Figure 6 depicts the expression level of TH, α-synuclein, PARP1, Bcl-2, Bax, and cleaved caspase-3 in the tissue lysate of SNpc using the western blot analysis. The expression of TH (Figure 6(a)) and Bcl-2 (Figure 6(d)) was observed to be significantly reduced (p < 0.001) in the MPTP-intoxicated group in comparison to the control group. However, the expression of TH (Figure 6(a)) and Bcl-2 (Figure 6(d)) was found to be significantly enhanced (p < 0.01)) in the MOE pre-treated group in comparison to the MPTP-intoxicated group, signifying the ameliorating effect of the MO leaf extract.
Evaluation of the Relative Expression of (a) Tyrosine Hydroxylase, (b) α-Synuclein, (c) PARP1, (d) Bax/Bcl-2 Ratio, and (e) Caspase-3 in the Tissue Lysate of the SNpc Region of the Mid-brain: Pre-treatment with the MOE Extract Imparts a Neuroprotective Effect After MPTP Intoxication by Ameliorating the Expression Levels of the Bax/Bcl-2 Ratio, Caspase-3, PARP1, α-Synuclein, and Tyrosine Hydroxylase. MOE Treatment Downregulated the Expression of Bax, Caspase-3, PARP1, and α-Synuclein, and Upregulated the Expression Level of Bcl-2 and TH Compared to the Diseased Group. The Values Are Expressed in Terms of Mean ± SEM (n = 5). *p < 0.05; **p < 0.01, and ***p < 0.001. The Results Were Analyzed via One-way ANOVA Followed by the Newman–Keuls Test Using the GraphPad Prism Software.
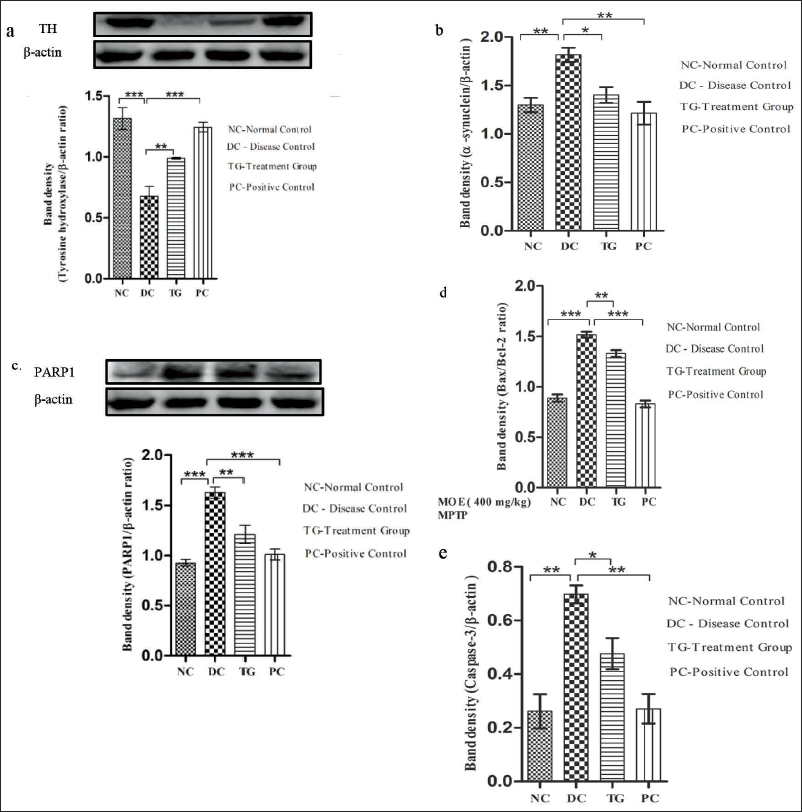
On the other hand, the expression of α-synuclein (p < 0.01) (Figure 6(b)), PARP1 (p < 0.001) (Figure 6(c)), Bax (p < 0.001) (Figure 6(d)), and cleaved caspase-3 (p < 0.01) (Figure 6(e)) was found to be significantly enhanced in the MPTP-intoxicated group in comparison to the saline-treated control group. However, the expression of α-synuclein (p < 0.05) (Figure 6(b)), PARP1 (p < 0.01) (Figure 6(c)), Bax (p < 0.01) (Figure 6(d)), and cleaved caspase-3 (p < 0.05) (Figure 6(e)) was found to be comparatively reduced in the MOE pre-treated group in comparison to the MPTP-intoxicated group, signifying the ameliorating potential of the MO leaf extract.
Discussion
The results from this study demonstrate that PARP1 inhibition could be a potential target for the therapeutic treatment of PD. In addition, the ethanolic extract of MO leaves could be used as a potential therapeutic agent to prevent the progression of the disease. The key findings of this study are as follows: (a) the bioactive compounds present in MO leaves could be a potent inhibitor of PARP1, as per the results of in silico docking analysis; (b) the MO leaf extract modulates MPTP-induced neurobehavioral deficits in mice; (c) the MO leaf extract modulates MPTP-induced changes in the mitochondrial complex I, II, IV, and V activity; (d) the MO leaf extract modulates MPTP-induced changes in the levels of mitochondrial superoxide dismutase and glutathione; and (e) the MO leaf extract modulates MPTP-induced changes in the expression of TH, α-synuclein, PARP1, and mitochondrial dysfunction–induced apoptotic pathway.
Kam et al. in their previous research have shown that the over-activated PARP1 protein can cause mitochondrial dysfunction and can change α-synuclein into more toxic forms, thus increasing the degenerative progression of PD. 3 PARP1 inhibition thus represents a potential therapeutic strategy for preventing the disease progression.
MO, a traditional herb from the Moringaceae family, is commonly recognized as the horse-radish tree, drumstick, or sahajna. It is widely used in lower-middle income nations as a source of nourishment for both humans and animals, primarily due to its established antioxidant, anti-inflammatory, and anti-apoptotic properties. These effects are attributed to various phytoconstituents present in MO, such as β-carotene, quercetin, kaempferol, ascorbic acid, flavonoids, phenolic acid, rhamnose, glycosylates, glucomoringin, and isothiocyanates. Research has indicated that MO contains numerous bioactive compounds that have favorable impacts on human health.18, 19
The data obtained in this study after in silico docking analysis hence suggested that the bioactive compounds present in the MO leaf extract could potentially inhibit the over-activated PARP1 protein.
The isothiocyanate isolated from the MO seed extract was found to be effective in modulating MPTP-induced behavioral deficits in a Parkinsonian mouse model. 17 The methanolic extract of MO leaf powder was reported to improve homocysteine-induced behavioral deficits in Alzheimer’s and cognitively impaired mouse model. 20 The findings of this study suggested that the ethanolic extract of MO leaf powder was effective in modulating MPTP-induced behavioral deficits in a Parkinsonian mouse model, which is in accordance with the previously reported findings.
Mitochondria play a fundamental role in the generation and regulation of the bioenergetics of cells that produce ATP molecules through oxidative phosphorylation. These functions are associated with neurodevelopment, connectivity, plasticity, and differentiation. 14 Mitochondrial dysfunction and oxidative distress are the key factors responsible for PD pathology. 21 Complex I inhibition by toxic substances can lead to the etiology of neurodegenerative diseases, and complex I inhibition initiates a self-enhancing cycle of events that reduce the ATP output and reactive oxygen species (ROS) elevation and promote mitochondrial dysfunction. 22 Antioxidants have been investigated for their effects on reduced mitochondrial dysfunction and improved development of specific bioactive chemicals in mitochondria using both in vivo and in vitro models.23, 24 According to the study outcome, MPTP led to mitochondrial dysfunction in mice brains, which is supported by the activity of the mitochondrial NADH dehydrogenase and ATPase enzymes being inhibited. Additionally, the activity of complex II and IV of ETS was also observed to be reduced.
One of the crucial elements that controls the formation of free radicals is the ETC driven by mitochondrial complex II.25, 11 There is significant evidence that MPTP intoxication contributes to mitochondrial dysfunction. 26 In our study, we found significant increase in mitochondrial oxidative damage in terms of a decrease in the levels of anti-oxidative enzymes Mn-SOD and mt-GSH in MPTP-intoxicated mice, which was found to be in accordance with an earlier study. 27 Mn-SOD is essential for adequate antioxidant action, as mice lacking it incur neurotoxicity, cardiac damage, and perinatal mortality.28, 29 Davey et al. postulated that the lower activity of complex I is caused by a decrease in the level of GSH in PC12 cells, which also results in the loss of the threshold effect. 30 The mechanism underlying complex I threshold hindered by the GSH level remains unknown. GSH is an antioxidant that has been shown to protect the mitochondria against lipid peroxidation. 31 In our study, MOE was found to be successful in lowering the burden on the mitochondrial antioxidative defense system by decreasing the formation of ROS, as seen by the increased levels of decreased mt-GSH and Mn-SOD. Because reduced glutathione is known to be the main defense of mitochondrial processes, the ability of MOE to improve the activities of the complexes of ETC may be attributable to the increased availability of this antioxidant.
In addition to energy production and regulation of free radicles, mitochondria effectively regulate the process of cell apoptosis and cell survival, which are considered as crucial elements of aging. The cascade of events leading to cell death is triggered by pathological changes by the leakage of cytochrome c and numerous pro-apoptotic chemicals from the mitochondria into the cytoplasm as a result of the mitochondrial membrane permeation. Although Bcl-2 and Bax are members of the same Bcl-2 family and have conflicting activities, the regulation of caspase activation is kept in check by the balance between the expression levels of pro-apoptotic and pro-survival proteins. 32 Bax demonstrates the pro-apoptotic function, whereas Bcl-2 is known for its anti-apoptotic action. Numerous studies have demonstrated that the neurotoxin MPTP disrupts the Bax/Bcl-2 equilibrium, increasing the activity of caspase-3 in dopaminergic neurons. 33 The activation of caspase-3, which results in the loss of membrane integrity caused by the activity of Bax and ultimately causes cell death, is mediated by the release of apoptogenic proteins such as cytochrome c into the cytosol from mitochondria. Studies conducted both in vivo and in vitro have revealed that MPTP poisoning causes caspase-3 to express itself more frequently. Upon the release of cytochrome c into the cytosol following mitochondrial dysfunction, caspase-9 activation causes the beginning of the apoptotic cascade. As a result, the morphology of the mitochondria changes, activating caspase-3 and causing apoptosis. 34 In our study, the Bax/Bcl-2 equilibrium was found to be disrupted in addition to the increased expression of caspase-3 in the MPTP-intoxicated group. Treatment with MOE restored the balance between Bax and Bcl-2 and ameliorated the expression of caspase-3, which may be attributable to the neuroprotective property of MO leaves.
One of the most crucial components in the biosynthesis of catecholamine, including dopamine, is TH, which is the rate-limiting enzyme. The development of PD depends greatly on changes in the TH expression or activity because it has a major impact on the synthesis of dopamine. 35 MPTP is used in mice to simulate the symptoms of PD, such as the loss of dopaminergic neurons in the substantia nigra pars compacta and striatal dopamine depletion. 36 In our study, the expression of TH was found to be reduced in the MPTP-intoxicated group. MOE was found to restore the level of TH in the treated group. MPTP induces the aggregation of α-synuclein in degenerating neurons of the substantia nigra. The redistribution of α-synuclein from its usual synaptic and axonal position to cell bodies and dendrites associated with MPTP toxicity is strikingly similar to the redistribution of α-synuclein reported in idiopathic PD and dementia with Lewy bodies.37, 38 In a study conducted by Zhang et al., the levels of α-synuclein aggregation were found to be higher in MPTP-intoxicated subacute Parkinson’s mice, 39 and our study showed similar results. Pathologic α-synuclein is known to activate PARP1 and accelerate PAR synthesis in addition to α-synuclein aggregation, leading to parthanatos. 3 In our study, the expression of both PARP1 and α-synuclein was found to be higher in MPTP-intoxicated mice, which was observed to be reduced after MOE treatment, thus suggesting the neuroprotective property of MO leaves.
Conclusion
The present study demonstrates that the MO leaf extracts have significant antioxidant and neuroprotective activity and can be used as a potent neuroprotective agent to inhibit the progression of PD by regulating the apoptotic pathway. Furthermore, MO leaf extract could be a potent inhibitor of PARP1, one of the factors inducing apoptosis. MO effectively ameliorates mitochondrial complexes that were significantly disturbed after MPTP intoxication. Mitochondrial antioxidants (SOD and GSH) were significantly restored by MOE in the MPTP-intoxicated mouse model of PD. The neuroprotective mechanism of MOE in PD is thus better understood as a result of this study. Further investigation is required to establish the safety and efficacy of MOE in preventing the progression of PD, which could be further explored for clinical interventions.
Footnotes
Acknowledgement
The authors are thankful to the Head of the Department of Biochemistry, Institute of Sciences, Banaras Hindu University (BHU), for providing the basic departmental facility and to the Interdisciplinary School of Life Sciences (ISLS), BHU, for their central facility.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to research, authorship and/or publication of this article.
Ethical Approval
Ethical approval was obtained from the Institutional Ethical Committee (Clearance Number: BHU/DoZ/IAEC/2021-2022/032), Department of Zoology, Institute of Sciences, BHU, Varanasi, India.
Funding
The authors received no financial support for the research, authorship and/or publication of this article.
Informed Consent
Not applicable