
Abstract
Objective
To test the selectivity and degree of functional agonism of Ocelot Bio’s dual agonist/antagonist molecule, OCE-205, at the vasopressin 1a receptor (V1aR).
Methods
Cells expressing human (h) or rat V1a, V1b, V2, or oxytocin receptors (OTR) were incubated with varying concentrations of OCE-205 or with arginine vasopressin (AVP), and responses were measured with fluorescence or reporter gene assays. In addition, human resistance arteries were exposed to increasing concentrations of OCE-205, and the resulting contractility was measured.
Results
The mean efficacy of OCE-205 at hV1aR was 39% of the maximal possible effect (MPE), with a mean EC50 of 0.71 nM. Above 1 nM OCE-205, the percent maximal possible effect (%MPE) plateaued. The EC50 was much higher at hV1bR (134 nM), hV2R (420 nM), and OTR (6.9 nM), indicating selectivity for hV1aR. Results at rat receptors were similar. OCE-205 produced 40.0% of maximal depolarization-induced contraction, demonstrating functional partial agonism.
Conclusion
The dual agonist/antagonist structure of OCE-205 thus allows it to act as a highly selective partial agonist at vasopressin V1aR at therapeutically relevant concentrations.
Introduction
New therapeutic alternatives are needed for decompensated cirrhosis and its associated hemodynamic complications. Portal hypertension due to cirrhosis leads to vasodilation of the splanchnic arteries and reductions in systemic vascular resistance, effective arterial blood volume, and arterial pressure. 1 In response, the kidney activates the renin-angiotensin-aldosterone system, causing retention of sodium and water. The non-osmotic release of vasopressin further increases water retention. These changes lead to intense intrarenal vasoconstriction that, together with reduced perfusion pressure, can cause renal hypoperfusion and hepatorenal syndrome-acute kidney injury (HRS-AKI), among other complications.1,2
Current treatment of HRS-AKI is based on the expansion of blood volume (e.g., with albumin) and short-term treatment with systemic vasoconstrictors to reverse renal dysfunction, with the long-term goal of liver transplantation. 3 The best-characterized vasoconstrictors for this purpose are vasopressin agonists. The primary function of vasopressin, stimulation of water absorption and retention, is mediated by V2 receptors (V2R), which are expressed in the distal tubules and collecting ducts of the kidney.4–6 At high physiologic and pharmacologic concentrations, vasopressin activates the vasopressin V1a receptor (V1aR), which is expressed on smooth muscle cells in the walls of arteries and veins and causes systemic vasoconstriction, 4 including of the splanchnic circulation. 2 Vasopressin also acts on the V1b receptor (V1bR), whose activation stimulates corticotropin secretion, ultimately leading to fluid retention. 7 A second hormone in the vasopressin system, oxytocin (OT), mediates labor and lactation through oxytocin receptors (OTR) expressed in the uterus and mammary gland, respectively.
Drugs used to treat HRS-AKI include norepinephrine; midodrine (an α-adrenergic agonist) and octreotide (a vasoconstrictor that mimics somatostatin), used alone or in combination;8,9 and, most recently, terlipressin. Alpha-adrenergic agonists such as norepinephrine and midodrine increase systemic vascular resistance, but the administration of norepinephrine requires a central venous line, often requiring intensive care. 3 Terlipressin has been used outside the USA for >10 years and received FDA approval in September 2022 to treat adults with HRS and rapid reductions in kidney function. Terlipressin is a prodrug of lysine vasopressin (LVP, the porcine version of arginine vasopressin (AVP)), which has activity at V1aR, V1bR, and V2R.10,11 Its metabolite, LVP, is a full V1a agonist that also maximally activates V2R at therapeutic concentrations. 11
Terlipressin, although recommended as first-line treatment for HRS-AKI,3,12 can cause significant adverse effects such as tissue hypoperfusion and ischemia—likely because of excessive vasoconstriction due to the full agonism of V1aR by LVP13–15—and fluid overload and respiratory failure via its stimulation of V2R, leading to sodium and water retention.10,16–19
To improve the safety profile of vasoconstriction through the vasopressin system in patients with HRS-AKI, a compound with high V1aR selectivity and less-than-maximal agonism of V1aR is needed. In 1996, Zhu proposed that a single molecule possessing both agonist and antagonist properties would act functionally as a partial agonist. 20 Ocelot Bio’s OCE-205 was designed as such a dual-action molecule, with two domains: one that can bind V1aR as an agonist and another that binds it as an antagonist. Here, we examined the cellular response to OCE-205 binding to rat and human V1aR and its effect on arterial contractility to determine whether, as predicted, it acts as a partial V1aR agonist. We also examined its specificity for V1aR relative to other vasopressin receptors.
Materials and Methods
Cell Lines
Studies were conducted with cell lines expressing rat (r) or human (h) V1a, V1b, V2, or OT receptors.
For the experiments with human receptors, human embryonic kidney (HEK)-flp-in cells (Invitrogen, Waltham, MA, USA) stably expressing the lacZ-Zeocin™ fusion gene were used for expression of hV1aR and hV1bR. These cells were designed for use with the Flp-ln™ expression vector 21 containing the gene of interest (here, hV1aR or hV1bR) and the Flp recombinase expression plasmid, pOG44. For hV2R and hOTR, HEK-293 cells (ATCC, Manassas, VA, USA) transiently expressing hV2R (Ferring Research Institute [FRI], Inc., San Diego, CA, USA) and CHO-K1 cells stably expressing hOTR (FRI, Inc.) were used.
For experiments with rat receptors, A7r5 rat thoracic aorta smooth muscle cells endogenously expressing rV1aR (ATCC), FLP-In 293 (HEK-293) cells (Invitrogen) stably expressing rV1bR (FRI, Inc.), HEK-293 cells (ATCC) transiently transfected with rV2 (FRI, Inc.), and Chinese hamster ovary (CHO)-K1 cells (ATCC) transiently expressing rOTR (FRI, Inc.) were used.
Cell Maintenance
HEK-flp-in cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM; Mediatech, Manassas, VA, USA) containing 10% (v/v) heat-inactivated fetal bovine serum (FBS; Invitrogen), 4 mM GlutaMAX™-I (Invitrogen), and 25 µg/mL hygromycin B at 37°C under 5% CO2 in a humidified atmosphere. The culture medium for hV1bR-expressing cells also contained 100 U/mL penicillin and 100 µg/mL streptomycin.
HEK-293 cells transiently expressing hV2R were maintained in DMEM containing 10% (v/v) heat-inactivated FBS and 4 mM L-glutamine (Mediatech) or GlutaMAX-I at 37°C under 5% CO2 in a humidified atmosphere.
CHO-K1 cells stably expressing hOTR were maintained in DMEM-F12 (Invitrogen) containing 5% (v/v) heat-inactivated FBS, 2 mM L-glutamine or GlutaMAX-I, and 900 µg/mL G418 sulfate at 37°C under 5% CO2 in a humidified atmosphere.
A7r5 cells were maintained in DMEM containing 10% (v/v) heat-inactivated FBS and 4 mM GlutaMAX-1 at 3°C under 5% CO2 in a humidified atmosphere.
On the day prior to the assay, cells were removed from culture flasks using trypsin EDTA, harvested in the medium used for cell culture, and seeded into 384-well (for V1a) or 96-well (for other receptors) poly-d-lysine-treated plates at 7.5 × 10 4 cells in 20 µL/well for rV1a, 2.5 × 10 4 cells in 20 µL/well for hV1a, and 4–5 × 10 4 cells in 100 µL/well for all other receptors.
Test Compounds
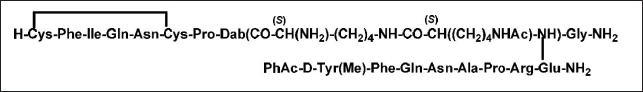
OCE-205 is a 20-mer monocyclic, branched peptide containing naturally and non-naturally occurring amino acids. The full peptide sequence is shown in Figure 1. OCE-205 (97.3% peptide purity) and AVP (reference agonist) were used in the functional cell-based assays. For all of these assays, compounds were prepared in 100% dimethyl sulfoxide (DMSO) at 10 mM stock concentrations (or 5 mM for AVP), stored at −20°C, and allowed to thaw just before the assay. The compounds were serially diluted to 10× working solutions in cell media. Blanks consisting of dilution media supplemented with 0.1% (v/v) DMSO were also used as controls in each study. No inhibitory effect of DMSO was seen at 0.1%.
For the contractility assay, OCE-205 was formulated as a 23.5 µM stock solution in physiological salt solution (PSS; 120 mM NaCI, 4.6 mM KCI, 1.5 mM NaH2PO4·1H20, 0.7 mM Na2HPO4, 11.5 mM D-glucose, 25 mM NaHCO3, 2.4 mM CaCI2, 1.2 mM MgCI2 [pH 7.35–7.45]). The stock solution was serially diluted in PSS to concentrations allowing for a further 1/100 dilution upon addition of the compound to the test apparatus in a cumulative fashion from lowest concentration to highest to obtain the final test concentration.
Functional Cell-based Assays
To detect activity generated by binding of the test compounds to endogenous rV1aR or the stably expressed hV1aR, fluorometric imaging plate reader calcium assays (Molecular Devices, San Jose, CA, USA) were performed in accordance with the manufacturer’s instructions. 22 Briefly, the real-time fluorescence of an intracellular calcium-sensitive dye was measured immediately upon the addition of the test compound at various concentrations. The endogenous ligand of V1aR, AVP, was used as the reference agonist.
Reporter gene assays were used to monitor agonist-induced activity at the human and rat V1b, V2, and OT receptors. Cells expressing the receptor of interest were transiently transfected with a luciferase reporter gene under the control of transcriptional regulatory elements responsive to receptor activation. One to two days after the transfection, the cells were incubated with OCE-205 at concentrations from 0.1 pM to 10 µM or a control for 5 h, and then the expression of the luciferase reporter gene was measured using the Luclite Reporter Gene Assay System (Perkin Elmer). As reference agonists, AVP was used in V1bR assays, desmopressin (dDAVP) in V2R assays, and carbetocin in the OTR assays.
For V1aR response, the area under the curve of the real-time calcium traces, expressed as relative fluorescence units, was determined. For V1b, V2, and OT receptor responses, luciferase activity was expressed in luminescent counts per second.
Compound potency was expressed as the concentration that produced a half-maximal response (EC50), calculated with a four-parameter non-linear regression analysis of concentration–response curves in ActivityBase™ software (IDBS, UK).
Efficacy was expressed in relative terms as the percent maximal possible effect (%MPE) relative to the maximal response of the reference agonist for each assay (AVP for V1aR and V1bR, dDAVP for V2R, and carbetocin for OTR). Results are expressed as the mean and 95% confidence interval unless otherwise stated.
Arterial Contractility Assay
Resistance arteries were isolated from human mesenteric tissue and finely dissected under a stereomicroscope while bathed in carbogen-aerated PSS maintained at 37°C. Arterial segments (2 mm) were mounted in a pressure myograph system between two glass cannulae in 7-mL tissue baths containing aerated PSS. Arteries were gradually pressurized to 60 mmHg (Figure 2). Arterial contractility was stabilized by potassium-induced depolarization with three consecutive exposures to a solution with a high potassium concentration (PSS with 124.34 mM potassium chloride (KCl) and no NaCl; 124K+PSS), each followed by a wash of the bath with aerated PSS. 23
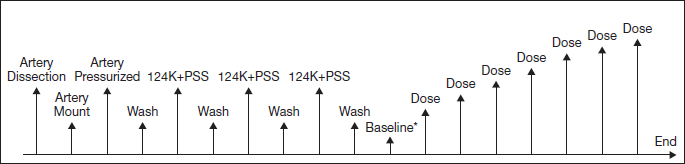
Cumulative concentration–response curves (CCRC) were then generated for the compounds. Arteries were bathed in aerated PSS containing the initial compound concentration (0.1 nM OCE-205), and each following dose was added to the bath without draining, taking into account the amount of compound already in the chamber when calculating the final concentration. The concentrations used were 0.1, 0.3, 1, 3, 10, 32, and 100 nM OCE-205. Only a single CCRC was generated in each arterial segment tested.
Contractile activity was determined by measuring the arterial outer diameter via digital video edge detection in response to each 124K+PSS depolarization and each compound concentration. Throughout the experiment, vessel diameter data were collected continuously using DMT Vessel Acquisition Suite software (DMT-USA, Ann Arbor, MI). For each 124K+PSS stimulation or compound concentration, data were collected until the vessel diameter was judged to have reached a plateau before proceeding to the next experimental step.
The vessel diameters for each artery preparation (in millimeters) were analyzed using a Microsoft Excel template. Data collected in response to the three 124K+PSS depolarization cycles were first used to determine artery stability. The contraction induced by the last stimulation (3rd 124K+PSS) was then used as the internal reference response of each artery (defined for the purposes of this study as 100% contraction) for reporting contractile activity. Arteries that could not be stabilized with three cycles of depolarization-induced contraction were not further examined.
Results
Functional Cell-based Assays
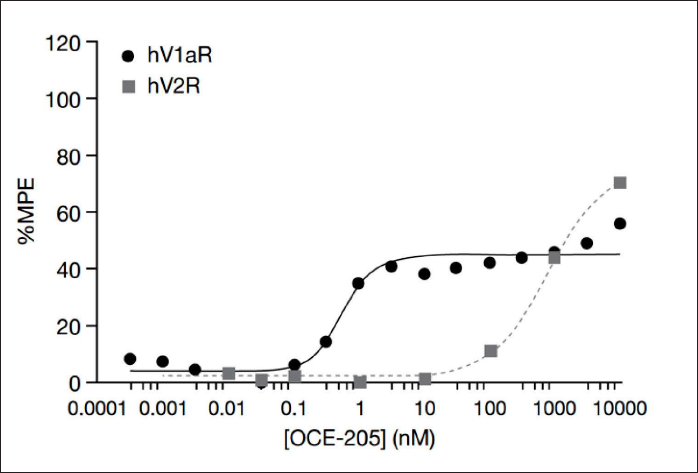
OCE-205 was a potent partial agonist at hV1aR in a cell-based calcium assay (EC50 = 0.71 nM, 39 %MPE; Table 1). The %MPE plateaued at a concentration of ~1 nM OCE-205, with no further increases in %MPE in response to further increases in OCE-205 concentration (Figure 3).
In Vitro Agonist Activities at Human Vasopressin Receptors.
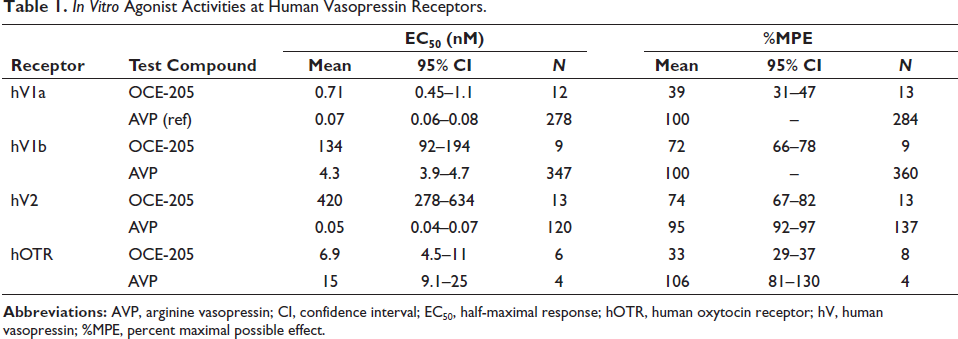
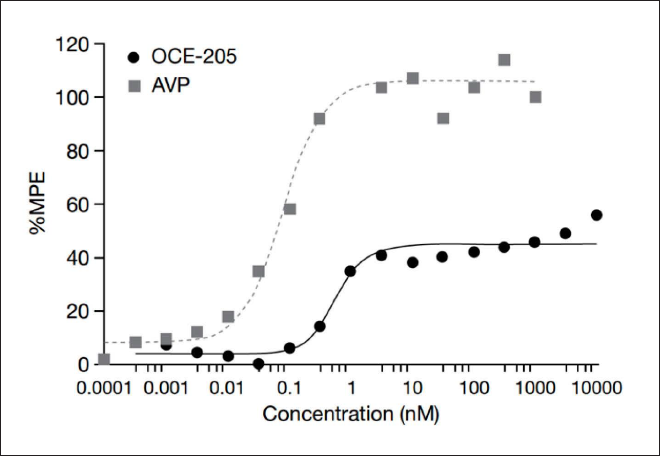
In addition, OCE-205 was functionally selective for hV1aR, displaying significantly lower potency at hV1bR (EC50 = 134 nM), hV2R (EC50 = 420 nM), and hOTR (EC50 = 6.9 nM) in cell-based reporter gene assays (selectivity ratios of 1:188:592:10; Table 1 and Figure 4). AVP was functionally a nonselective agonist at the human V1a, V1b, V2, and OT receptors, with EC50 values of 0.07, 4.3, 0.05, and 15 nM, respectively. OCE-205 was also a potent partial agonist at rV1aR in the cell-based calcium assay (EC50 = 0.22 nM, 30 %MPE; Table 2). Consistent with the results at the human receptors, OCE-205 was functionally selective for rV1aR over rV2R, and AVP was a nonselective agonist at the rat receptors tested (Table 2).
In Vitro Agonist Activities at Rat Vasopressin Receptors.
Arterial Contractility Assay
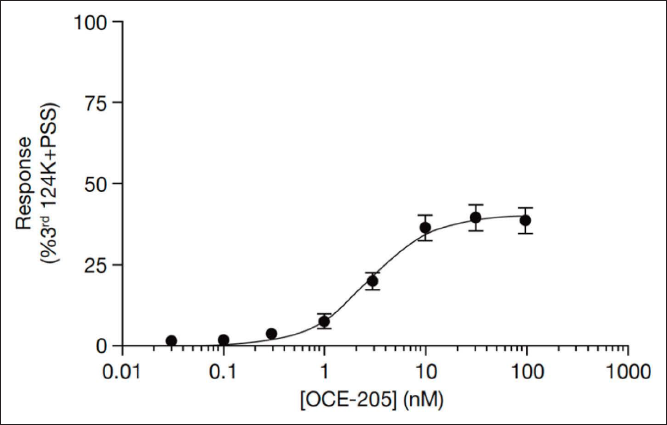
Treatment of human mesenteric resistance arteries with OCE-205 resulted in an attenuated maximal response (40.0%) relative to the maximal depolarization achieved with potassium (Figure 5).
Discussion
OCE-205’s unique design of vasopressin V1aR agonist and antagonist domains within a single molecule provided a mean response of 39 %MPE in HEK cells expressing hV1aR, indicating that, as predicted, OCE-205 effectively acts as a partial agonist of V1aR. Confirming this, treatment of human mesenteric resistance arteries with OCE-205 resulted in an attenuated maximal response (40.0%) relative to the third stimulation with potassium, set to 100%, which is expected to be close to the contraction elicited by the endogenous ligand, AVP. 24 The translation of OCE-205 V1aR partial agonism from in vitro cell-based responses to ex vivo responses in mesenteric vessels is consistent with the concept that treatment with OCE-205 would result in submaximal vasoconstriction of the splanchnic vasculature in patients with portal hypertension, thereby reducing portal blood flow and pressure and improving the systemic hemodynamics of patients with HRS-AKI, 1 but with a lower risk of ischemia than a full V1aR agonist.
Furthermore, OCE-205 was selective for V1aR at a wide range of concentrations. Although it elicited a higher maximal response at V2R (74 %MPE) than at V1aR (39 %MPE), this occurred only at concentrations about 1,000 times higher than the lowest concentration that elicited the maximal response at V1aR (Figure 3), where no V2R activity was seen. Thus, at clinically relevant concentrations, OCE-205 has little to no activity at human V2R and, unlike existing vasopressin agonists, is therefore unlikely to cause further water retention and its clinical sequelae due to V2R activation.
Acute HRS-AKI is defined by recent guidelines as an increase in serum creatinine of ≥0.3 mg/dL within 48 h, an increase in serum creatinine of ≥1.5 times baseline, or urine volume <0.5 mL/kg/h for 6 h. 25 Patients with HRS-AKI have a poor prognosis, with fewer than half of patients surviving 90 days. 26 Because the prevalence of decompensated cirrhosis is increasing, 27 the incidence of HRS-AKI and the need for timely treatment are also likely to increase. Furthermore, OCE-205 has the potential for more generalized use for other complications of decompensated cirrhosis, such as resistant and refractory ascites or post-paracentesis–induced circulatory dysfunction, because the underlying mechanisms of renal dysfunction are similar.28–30
Terlipressin is effective at improving renal function in patients with HRS-AKI,13,31–33 but it has also been associated with ischemia, fluid overload, and other adverse events due to its nonselective effects on both V1a and V2 receptors.13–15 Because of its innovative design, OCE-205 binds to a given receptor at either its agonist domain or its antagonist domain. Therefore, over a population of OCE-205–occupied vasopressin receptors, a fraction will be occupied by the agonist domain while another fraction will be occupied by the antagonist domain, resulting in effective partial agonism of the receptor and limiting the maximum achievable vasoconstriction. This essentially eliminates the adverse effects due to excessive vasoconstriction seen with full agonists of the V1aR.
The conclusions that can be drawn from this study are limited by its in vitro nature; however, the data reported here show that OCE-205 is a promising candidate for in vivo studies to counteract HRS-AKI and ascites. In particular, because we have shown that OCE-205 has similar effects on rat and human vasopressin receptors, the rat can be used as a model for further studies of its mechanism and in vivo effects. Results from preclinical in vivo studies are in preparation, and a phase 2 clinical trial (NCT05309200) is currently in progress. If successful, OCE-205 could represent an important new therapeutic for the treatment of HRS-AKI and other conditions resulting from portal hypertensive complications of cirrhosis.
Conclusion
The dual agonist/antagonist structure of OCE-205 allows it to act as a functional partial agonist at vasopressin V1aR, achieving a maximal response of about half that achieved by the endogenous ligand, AVP, at concentrations likely to be therapeutic. In addition, it is highly selective for V1aR, suggesting that it will not produce undesired water retention through the activation of V2R. OCE-205 is therefore an ideal candidate for further evaluation as a treatment for HRS-AKI and ascites.
Footnotes
Abbreviations
AKI, acute kidney injury; AVP, arginine vasopressin; CHO, Chinese hamster ovary; CCRC, cumulative concentration–response curve; Dab, 2,4-diamino butyric acid; dDAVP, desmopressin; DMEM, Dulbecco’s modified Eagle’s medium; DMSO, dimethyl sulfoxide; D-Tyr(Me), O-methyl-
Acknowledgments
Medical writing and editorial assistance were provided by Innovative Strategic Communications (Milford, PA) and funded by Ocelot Bio, Inc. We thank David Bernstein (Ocelot Bio) for his comments on the manuscript.
Data Availability
All relevant data are included in the manuscript. The original datasets cannot be shared because they are proprietary.
Declaration of Conflicting Interests
S.B. is an employee and founder of Ocelot Bio, Inc. G.H. is a founder of and consultant to Ocelot Bio, Inc. G.C., E.E.C., J.T., H.T., R.L., and G.H. were employees of Ferring Pharmaceuticals, Inc. at the time of the study.
Ethical Statement
Ethics committee approval and patient consent were not required for this in vitro study.
Funding
The original study was supported by Ferring Pharmaceuticals, Inc.