
Abstract
Background:
Down syndrome (DS) is a multisystem genetic condition requiring anticipatory, longitudinal, and development-centred care. Although survival has improved significantly, morbidity remains substantial due to congenital anomalies, neurodevelopmental disabilities, and acquired medical conditions.
Objective:
To synthesise current international and Indian guidelines and provide a practical, age-anticipatory clinical framework for the comprehensive management of children with DS.
Methods:
This review integrates evidence from established guidelines and contemporary literature, adopting a problem-oriented, age-based approach. It outlines the expected timeline of comorbidities, their clinical presentation, recommended investigations, and evidence-based management strategies.
Results:
A structured clinical framework is presented that emphasises early recognition, timely evaluation, and individualised interventions. Key domains include surveillance of systemic comorbidities and a strong focus on neurodevelopmental and behavioural assessment, identified as primary determinants oflong-term functional outcomes. The review also highlights context-specific considerations relevant to diverse healthcare settings.
Keywords
Introduction
Down syndrome (DS), resulting from trisomy 21, is the most common chromosomal aneuploidy compatible with long-term survival, occurring in approximately 1 in 700–800 live births worldwide.[1] In India, reported prevalence is variable and likely underestimated due to inconsistent prenatal screening and reporting practices.[2] Advances in cardiac surgery, neonatal care, infection control, and early intervention services have dramatically improved survival, with current life expectancy reaching 55–60 years.[3]
Accordingly, the focus of care has shifted from survival alone to anticipatory, longitudinal management, addressing medical comorbidities, neurodevelopmental trajectories, behavioural health, and transition to adult life. Recent guidelines from the American Academy of Paediatrics (AAP, 2022) and the Indian Academy of Paediatrics (IAP) emphasise structured surveillance combined with individualised, family-centred care.[4,5]
Epidemiology and Aetiology
DS arises from an extra copy of chromosome 21 dueto nondisjunction, translocation, or mosaicism. Sporadic nondisjunction accounts for ~95% of cases, translocationsfor ~3%–4%, and mosaicism for ~1%–2%. Parental age, primarily maternal, influences nondisjunction risk. Formal genetic counselling is recommended to discuss recurrence risks, which vary by aetiology.
Clinical Phenotype and Natural History
Children with DS demonstrate a distinct phenotype: hypotonia, epicanthal folds, flat nasal bridge, single transverse palmar crease, and joint laxity. Developmental profiles typically include intellectual disability of variable severity and delayed motor and communication milestones. DS also confers increased risks for multiple co-occurring medical conditions: congenital heart disease (CHD) (~40%–50%), hearing loss (~75%), vision disorders (~60%–80%), obstructive sleep apnoea (OSA) (~50%–79%), thyroid disease, and haematologic abnormalities. However, phenotypic expression is heterogeneous, and long-term outcomes are largely determined by the type, timing, and quality of management of associated comorbidities, rather than by the chromosomal diagnosis itself.[6]
Problem-oriented Management Across the Lifespan
Congenital Heart Disease
CHD affects approximately 40%–50% of children with DS and remains the most important predictor of early morbidity and mortality.[4,7] Atrioventricular septal defects (AVSD) account for 30%–45% of cases, followed by ventricular septal defects (25%–35%), atrial septal defects (15%–20%), and patent ductus arteriosus (5%–10%).[7] Pulmonary hypertension occurs in 5%–15% and may develop even in the absence of structural heart disease.[8] Clinical recognition in the neonatal period includes murmur, tachypnea, feeding difficulty, and poor weight gain, although some lesions, particularly AVSD, may present subtly initially. All neonates with DS should undergo echocardiography regardless of clinical findings [Table 1].[4] Early surgical repair, especially for complete AVSD within the first 6 months of life, has resulted in infant survival exceeding 95% in contemporary cohorts,[3,9] while delayed repair is associated with pulmonary hypertension and poorer long-term outcomes.
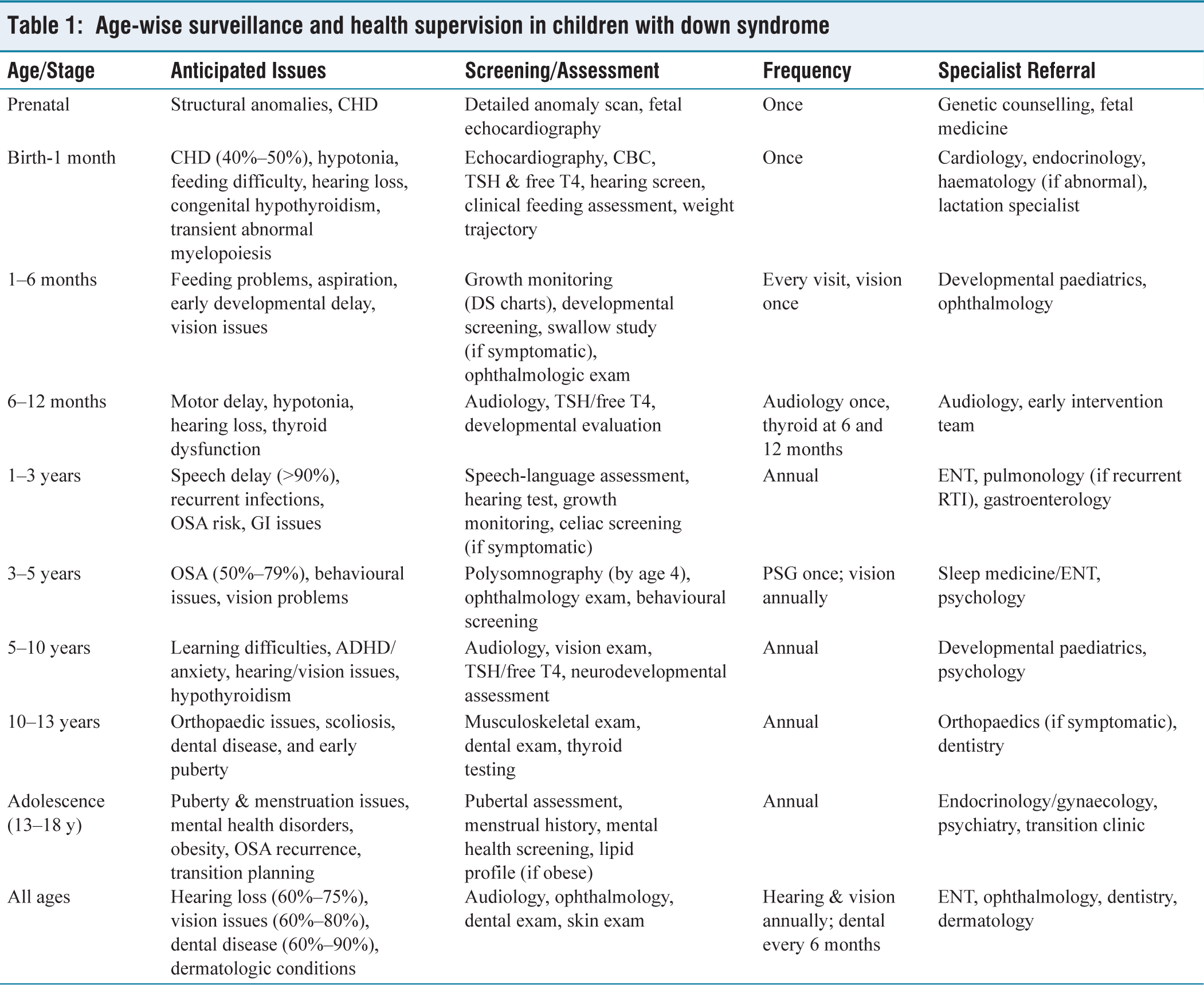
Age-wise surveillance and health supervision in children with down syndrome
Cognitive and Motor Development
More than 95% of individuals with DS have intellectual disability, most commonly mild to moderate (70%–85%), while 10%–15% have severe disability.[10] Motor delays are observed in 80%–90% of children, driven by hypotonia (>90%), ligamentous laxity, and delayed postural control.[11] Clinical recognition from infancy onward includes delayed head control, sitting and walking, as well as expressive language delays disproportionate to receptive skills. Developmental screening at every visit, formal evaluation at 6–12 month intervals, and adaptive functioning assessments (e.g., Vineland) are recommended to guide interventions. Early initiation of combined physiotherapy, occupational therapy, and intensive speech-language therapy has been shown to improve adaptive functioning and communication outcomes, even if IQ itself remains largely unchanged.[12]
Speech, Language and Communication Disorders
Speech and language delays affect over 90% of children with DS and represent the most significant functional limitation.[13] These challenges typically become apparent in the toddler and preschool years, with delayed first words, poor speech intelligibility, and frustration-related behaviours. Contributing factors include hypotonia, hearing loss, and oral motor dysfunction. Management involves early and sustained speech therapy, augmentative and alternative communication (AAC) when appropriate, and aggressive correction of hearing impairment. Language ability is a major predictor of social integration, independence, and school readiness.
Behavioural and Psychiatric Comorbidities
Behavioural and mental health disorders are common but frequently under-recognised in DS. ADHD-like symptoms occur in 20%–40%, anxiety disorders in 10%–30%, autism spectrum disorder in 5%–10%, and depression in 5%–15% of adolescents.[14,15] Early language delay often exacerbates behavioural challenges, which may manifest as frustration or attention difficulties. Behavioural regression should prompt evaluation for medical contributors such as hypothyroidism, OSA, hearing impairment, or psychosocial stressors before assigning a primary psychiatric diagnosis.[4,16]
Assessment and Management Strategies
Structured assessment includes baseline developmental evaluation by 12 months, annual tracking with standardised tools, and behaviour rating scales for ADHD and autism. Management is multifaceted, combining therapy, environmental adaptations, parental training, and school Individualised Education Plans (IEPs) starting as early as possible. Judicious pharmacotherapy may be indicated when behavioural challenges are severe. Neurodevelopmental prognosis is typically mild to moderate, with adaptive functioning and independence varying widely; language skills and behaviour are stronger predictors of adult independence than IQ.
Hearing and Vision: Modifiable Determinants of Development
Hearing and vision impairments are common and modifiable determinants of neurodevelopment in children with DS. Hearing loss affects 60%–75% of children, predominantly conductive (50%–70%) due to chronic otitis media, while sensorineural loss occurs in 10%–20%.[17] Visual abnormalities are observed in 60%–80% of children, including refractive errors (40%–60%), strabismus (20%–40%), and cataracts (5%–15%).[18] Annual audiologic and ophthalmologic surveillance from infancy onward is recommended to prevent secondary developmental delays and optimise functional outcomes [Table 1].[4]
Endocrine Disorders
Endocrine disorders, particularly thyroid dysfunction, are among the most frequent medical comorbidities in DS. Congenital hypothyroidism occurs in 2%–5%, while acquired hypothyroidism develops in 25%–40% during childhood and adolescence[19] Diabetes mellitus has a lifetime prevalence of 2%–5%, higher than in the general population.[20] Routine biochemical screening, including TSH and free T4 at birth,6 months, 12 months, and annually thereafter, is recommended, as even subclinical hypothyroidism can adversely affect growth, tone, and cognition.[4,5] Clinical recognition includes poor growth, lethargy, and worsening developmental trajectory, and management consists of standard levothyroxine therapy. Thyroid dysfunction can mimic expected developmental delays and should not be overlooked.
Sleep-disordered Breathing
OSA affects 50%–79% of children with DS, with 20%–30% experiencing moderate to severe disease.[21] Contributing factors include adenotonsillar hypertrophy, midface hypoplasia, hypotonia, and obesity, which collectively predispose to airway obstruction. Clinically, OSA may present with snoring, daytime sleepiness, and behavioural problems, and is increasingly recognised as a contributor to cognitive and behavioural dysfunction.[22] The AAP recommends polysomnography at least once by 4 years of age, regardless of symptoms.[4] Management typically involves adenotonsillectomy, with continuous positive airway pressure (CPAP) therapy if residual OSA persists, emphasising the importance of early recognition and intervention to mitigate neurocognitive and behavioural impacts.
Respiratory Morbidity: Practical Management Perspective
Children with DS are at increased risk of respiratory morbidity due to upper and lower airway anatomical differences, generalised hypotonia, immune dysregulation, and frequent coexisting CHD. Recurrent respiratory tract infections affect approximately 40%–60% of children, particularly during infancy and early childhood, and represent a major cause of hospitalisation and healthcare utilisation.[23,24]
Obstructive airway disease is common. Structural airway abnormalities such as laryngomalacia, tracheomalacia, and bronchomalacia contribute to persistent stridor, recurrent wheeze, and poor response to standard asthma therapies. Recurrent wheezing or asthma-like symptoms occur in 15%–30% of children and are often multifactorial, related to airway malacia, aspiration, gastroesophageal reflux, or chronic infection rather than true atopic asthma.[25]
Aspiration due to oropharyngeal dysphagia is an important and frequently under-recognised contributor to chronic respiratory symptoms. Objective swallow studies demonstrate aspiration or penetration in up to 30%–50% of symptomatic infants and young children with DS. Children with recurrent pneumonia, chronic cough, feeding difficulties, or failure to thrive should therefore undergo formal swallow evaluation.[26]
Pulmonary hypertension affects approximately 5%–15%of children with DS and may occur with or without CHD. Chronic hypoxia from untreated OSA, recurrent infections, and aspiration are important, potentially reversible contributors.[8]
Management priorities include ensuring complete routine and high-risk immunisations (including influenza and pneumococcal vaccines), maintaining a low threshold for evaluation of airway abnormalities and aspiration, prompt treatment of respiratory infections, and multidisciplinary coordination involving paediatrics, pulmonology, otolaryngology, cardiology, and speech-language therapy.[4,5] Early identification and management of reversible respiratory contributors significantly reduce morbidity and improve functional outcomes.
Gastrointestinal and Nutritional Issues
Feeding difficulties are common in infants with DS, affecting 30%–50%, with gastroesophageal reflux reported in20%–40%, duodenal atresia in 5%–8%, and Hirschsprung disease in 1%–2%.[27,28] Celiac disease occurs in 5%–10%.[29] Clinically, these infants may present with poor latch, prolonged feeds, choking, or failure to thrive despite adequate intake. Contributing factors include hypotonia, poor suck-swallow coordination, cardiac disease, and gastroesophageal reflux. Early feeding assessment, including clinical evaluation and videofluoroscopic swallow studies when aspiration is suspected, is essential. Management focuses on feeding therapy, texture modification, pacing strategies, and nutritional rehabilitation guided by DS-specific growth charts. Early recognition and intervention reduce growth faltering and alleviate parental stress.
Haematologic Disorders
Transient abnormal myelopoiesis occurs in 10%–20% of neonates with DS and typically resolves spontaneously; however, careful monitoring is required due to the risk of progression. The lifetime risk of leukaemia is approximately 1%–2%, with a strong association with acute megakaryoblastic leukaemia.[30,31] Clinically, affected infants may presentwith bruising, pallor, or hepatosplenomegaly. Management includes routine neonatal blood counts and prompt haematology referral if abnormalities are detected to ensure early identification and intervention.
Musculoskeletal Health
Musculoskeletal involvement is a near-universal feature of DS, with generalised hypotonia present in over 90% of children and ligamentous laxity affecting more than 80%, predisposing to delayed motor milestones and a spectrumof orthopaedic complications. Pes planus is observed in 60%–80% of individuals, scoliosis in approximately 10%–15%, and varying degrees of joint instability may limit functional mobility and endurance.[11,32] Atlanto-axial instability (AAI), defined radiologically by an increased atlanto-dens interval, is reported in 10%–30% of children with DS; however, clinically significant spinal cord compression is rare, occurring in fewer than 2%.[33,34] Musculoskeletal issues are present from infancy through adolescence and evolve with growth, with joint pain and overuse injuries, particularly related to sports and repetitive activities, increasingly recognised in older children and adolescents.
Guideline-aligned management emphasises clinical vigilance rather than routine radiologic screening, as cervical spine radiographs poorly predict neurological risk.[4,5] Instead, careful musculoskeletal and neurological examinationshould be performed at every visit, with prompt evaluation of red-flag symptoms such as neck pain, gait deterioration,limb weakness, hyperreflexia, or changes in bladder orbowel function. Gait and strength assessment, foot posture evaluation, and targeted imaging are recommended only when clinically indicated. Early physiotherapy focusing on core strength and motor skills, use of orthotics for symptomatic pes planus, and activity modification favouring low-impact sports (e.g., swimming, cycling) form the cornerstone of management. High-impact or contact sports should be avoided unless cleared by orthopaedics, and suspectedAAI warrants urgent referral with avoidance of neck hyperextension. With timely intervention, most individuals with DS remain ambulatory and achieve good long-term functional outcomes and quality of life.
Dermatological Conditions
Immune dysregulation associated with trisomy 21 contributes to a high prevalence of dermatological disorders in children with DS. Atopic dermatitis affects approximately 20%–40%, while seborrheic dermatitis is reported in 10%–20%; autoimmune skin conditions such as alopecia areata (5%–10%) and vitiligo (3%–10%) also occur with increased frequency.[35,36] Milia and delayed wound healing may be observed. Dermatological manifestations are typically recognised clinically by pruritic, lichenified eczematous lesions (often in flexural areas), patchy hair loss, or hypopigmented macules. Diagnosis is primarily clinical, with skin biopsy or allergy testing reserved for atypical presentations or refractory eczema. Management focuseson regular skin hydration, topical corticosteroids for inflammatory flares, antihistamines for pruritus, and early dermatology referral for severe or unusual disease. Although rarely life-threatening, chronic skin conditions can significantly affect comfort, sleep quality, self-esteem, and caregiver burden; however, with routine care and anticipatory management, most dermatologic conditions in DS are chronic but well-controlled.
Oral and Dental Health
Oral health issues are highly prevalent yet frequently under-addressed in children with DS. Periodontal disease affects approximately 60%–90% of adolescents and adults andmay begin as early as late childhood, while delayed tooth eruption occurs in over 50% and malocclusion in 50%–70%, contributing to feeding difficulties, speech articulation problems and psychosocial concerns.[37,38] Macroglossiaand challenges with oral hygiene further increase the riskof dental caries. Oral health concerns should be anticipated from infancy through adolescence and recognised clinically by poor dental alignment, gingival inflammation or bleeding, and plaque accumulation with food trapping. Preventive dental evaluation is recommended at least every 6 months, with panoramic radiography reserved for orthodontic planning when indicated. Management should emphasise early dental referral by 1 year of age, regular reinforcement of oral hygiene practices, fluoride application and sealants, and timely orthodontic intervention during school age,with consideration of special-needs dental sedation when cooperation is limited. Early and consistent preventivedental care, coupled with caregiver education and routine surveillance, significantly reduces long-term oral morbidity and improves feeding, speech outcomes and quality of life.
Puberty, Menstruation and Sexual Health
Pubertal development in adolescents with DS is typically normal in timing and progression in over 80% of individuals, although growth patterns and body composition may differ from those of peers. Menstrual irregularities are common in the early post-menarchal period, affecting approximately 30%–50% of adolescent girls, but usually stabilise over time; dysmenorrhoea and psychosocial concerns related to body image, privacy and autonomy may also be present.[39] Female fertility is reduced but well documented, while male fertility is rare, though sporadic cases have been reported.[40] Importantly, adolescents with DS experience typical sexual development and curiosity, underscoring the need for proactive, developmentally appropriate counselling. Clinical management should include menstrual education beginning in pre-adolescence, non-hormonal measures such as NSAIDs for mild dysmenorrhoea, and hormonal options (combined hormonal contraception or progestin-only regimens) for heavy or disruptive menstruation in consultation with gynaecology or endocrinology.
Evaluation of delayed puberty should include thyroid and sex hormone assessment, with pelvic ultrasonography reserved for suspected structural concerns. Sexuality and relationship health discussions should be age-appropriate, developmentally informed, family-inclusive, and respectful of safety, consent, and self-advocacy. Informed education and safeguarding conversations reduce vulnerability to exploitation and promote healthy relationships, autonomy and dignity, and with appropriate guidance and endocrine support, most girls with DS experience normal pubertal development and manageable menstruation.[5,41]
Surveillance and Health Supervision
Transition to Adult Care
As children with DS age, a structured transition from paediatric to adult healthcare becomes essential to maintain continuity of care and optimise long-term outcomes. Transition planning should begin in early adolescence, addressing medical, psychosocial, educational, and vocational needs. Adults with DS require ongoing surveillance for CHD sequelae, thyroid dysfunction, sleep apnoea, obesity, hearing and vision impairments, and mental health conditions [Table 1].[1] Regular engagement with primary care physicians, adult specialists, and multidisciplinary teams ensures preventive care, timely management of comorbidities, and support for autonomy and self-advocacy. Transition also involves education on sexuality, reproductive health, and independent living skills, tailored to cognitive and adaptive abilities,while actively involving families and caregivers. Effective transition programmes have been associated with improved adherence to care, reduced hospitalisation, and enhanced quality of life in adults with DS.
Factors Affecting Outcomes and Survival
Outcomes and prognostication in children with DS are influenced by multiple interrelated factors. Key determinants include the presence and severity of CHD, timely initiation of neurodevelopmental interventions, correction of hearingand vision impairments, family support, socio-economic circumstances, and access to inclusive education. Survival trends have improved markedly, with infant survival exceeding 95% in high-resource settings and median life expectancy now reaching 55–60 years. Early mortality, however, remains primarily related to cardiac complications and infectious causes, underscoring the importance of comprehensive, anticipatory, and multidisciplinary care.
Organisations such as DS International offer global advocacy, inclusion, and peer support networks, enabling individuals and families to connect, share experiences, and engage in collective empowerment.[8] In India, groups like the DS Federation of India provide counselling, therapy camps, skill development activities, and family support services, while grassroots organisations such as the DS Support Group India (Smiling Dandelion Foundation) create community networks for peer support, education, and inclusion. Such community resources, alongside mainstream mental health services and helplines offering counselling and emotional support nationwide, are valuable complements to clinical care for promoting mental well-being and sustained community participation.
Prenatal Counselling
Paediatricians should provide balanced, non-directive prenatal counselling when a prenatal diagnosis is made. Topics include the nature and variability of DS, potential outcomes, available treatments and interventions, support resources, and reproductive options. Genetic counselling should discuss recurrence risks and options for prenatal testing in future pregnancies.
Recurrence Risk and Prevention in Future Pregnancies
The recurrence risk of DS depends on the underlying genetic mechanism. For most cases caused by nondisjunction, the recurrence risk is low (approximately 1%) but increases with maternal age. Translocation-related cases carry a higher recurrence risk, particularly if a parent is a balanced carrier, which can be as high as 10%–15% depending on the typeof translocation.[1] Genetic counselling is recommendedfor all families, both to clarify recurrence risk and todiscuss reproductive options. Prenatal screening—including noninvasive prenatal testing (NIPT), chorionic villus sampling, or amniocentesis—can be offered in subsequent pregnancies, and preimplantation genetic testing (PGT) may be considered in assisted reproductive settings to reduce recurrence risk.
Conclusion
DS is best understood as a lifelong condition characterisedby predictable, manageable medical and developmental challenges. A proactive, prevalence-guided approach—integrating surveillance, early intervention, and family-centred care—optimises outcomes and enables children with DS to achieve their fullest potential.
Supplemental material
Supplemental material for this article is available online.
Footnotes
Acknowledgements
The author acknowledges the contribution of multidisciplinary paediatric teams and families of children with Down syndrome, whose lived experiences continue to inform best clinical practice.
Declaration of conflicting interests
The author declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Funding
The author received no financial support for the research, authorship and/or publication of this article.
Institutional ethical committee approval number
Not applicable. This manuscript is an invited narrative review and did not involve human participants or patient data.
Informed consent
Not applicable.
Credit author statement
Not applicable.
Data availability
Not applicable.
Use of artificial intelligence
Not applicable.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.