
Abstract
Background:
Hypokalemic paralysis is a reversible yet potentially life-threatening cause of acute flaccid weakness. While primary periodic paralysis is well recognised, secondary forms due to renal tubular dysfunction are often overlooked. Fanconi syndrome, a proximal tubulopathy, is an uncommon autoimmune manifestation of Sjögren’s syndrome, which more typically presents with distal renal tubular acidosis (RTA). Its occurrence in a 19-year-old young female is highly atypical and proximal tubular involvement in Sjögren’s is distinctly rare.
Case Summary:
We describe a 19-year-old female who presented with acute flaccid quadriparesis and severe hypokalaemia. Laboratory evaluation revealed hypophosphatemia, non-anion gap metabolic acidosis and inappropriately alkaline urine. Urinalysis showed phosphaturia, glucosuria and proteinuria, confirming Fanconi syndrome. Electromyography (EMG) demonstrated a myopathic pattern. Strongly positive Anti-Sjögren’s syndrome-related antigen A (Ro) antibody (anti-SSA/Ro) antibodies and polyclonal hypergammaglobulinemia suggested an autoimmune aetiology consistent with renal-limited Sjögren’s syndrome. Targeted correction of electrolytes, vitamin D deficiency and metabolic acidosis led to rapid and complete recovery. The combination of very young age and isolated proximal tubular dysfunction underscores the diagnostic challenge and highlights the importance of considering renal-limited autoimmune disease even without sicca symptoms.
Conclusion:
This case illustrates a rare presentation of autoimmune Fanconi syndrome manifesting as hypokalemic paralysis in a young female. Anti-SSA/Ro seropositivity and polyclonal hypergammaglobulinemia, despite absent sicca features, should prompt evaluation for renal-limited Sjögren’s syndrome. Early recognition is crucial, as timely metabolic correction results in complete clinical recovery.
Keywords
Introduction
Hypokalemic paralysis is a neurological emergency characterised by acute, flaccid and often symmetrical weakness caused by a rapid reduction in extracellular potassium. While primary forms such as familial hypokalemic periodic paralysis (HypoKPP) are well recognised, secondary causes, particularly renal tubular acidosis (RTA) and Fanconi syndrome, are frequently overlooked because of overlapping clinical presentations.[1,2]
Fanconi syndrome represents a global proximal tubular reabsorptive defect leading to urinary losses of potassium, phosphate, glucose, amino acids and bicarbonate. These abnormalities result in non-anion gap metabolic acidosis, hypophosphatemia and hypokalaemia, all of which contribute to acute neuromuscular weakness.[3,4] Early identification is crucial because management differs fundamentally from primary periodic paralysis and typically leads to complete recovery.
Although Sjögren’s syndrome most commonly presents with distal RTA, proximal tubular involvement causing Fanconi syndrome is rare and reported only in isolated cases. Its occurrence in a young adult, particularly without sicca symptoms, poses a significant diagnostic challenge and underscores the importance of considering renal-limited autoimmune tubulointerstitial disease even in atypical age groups.
We report a case of severe hypokalemic paralysis secondary to Fanconi syndrome in a young female, highlighting the metabolic, diagnostic and therapeutic complexities of this underrecognised presentation.
Case Presentation
A 19-year-old previously healthy female presented with a three-day history of progressive bilateral lower limb (LL) weakness, followed by involvement of the upper limbs (UL) over the subsequent two days. The weakness was symmetrical and predominantly proximal, with the LL muscles more severely affected than the UL. There was no associated sensory loss, cranial nerve involvement or bladder/bowel disturbance and deep tendon reflexes were preserved throughout.
On clinical examination, motor power in the LLs was graded 2/5 proximally and 3/5 distally, while in the UL it was 3–4/5 proximally and 4/5 distally. Reflexes were preserved in all limbs. Cranial nerves were intact and there were no signs of cerebellar dysfunction or autonomic involvement. Sensory examination was normal.
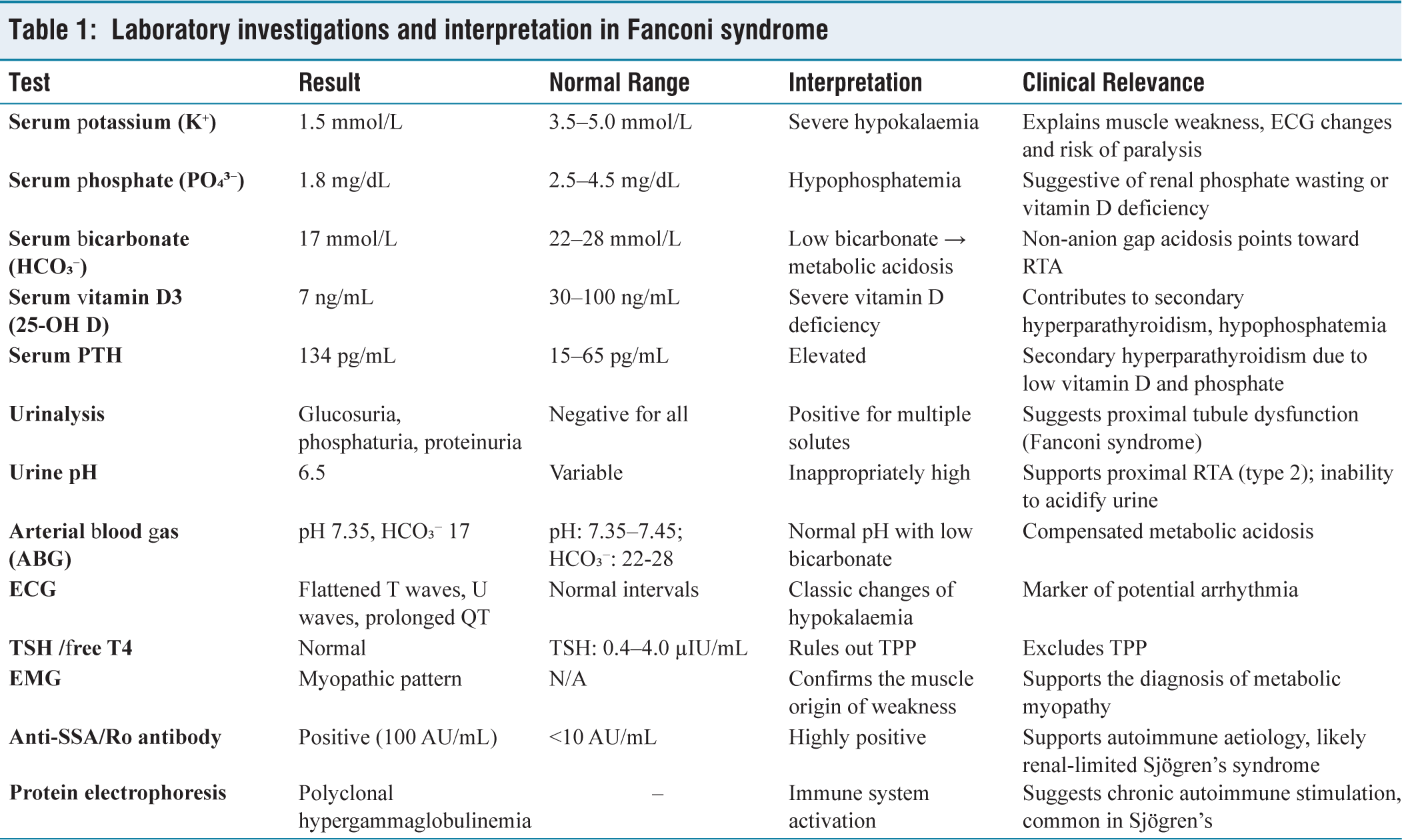
Initial laboratory investigations revealed severe hypokalaemia (serum potassium 1.5 mmol/L) and hypophosphatemia (1.8 mg/dL), along with low bicarbonate (17 mmol/L), consistent with a non-anion gap metabolic acidosis. Additional testing demonstrated low serum vitamin D3 (7 ng/mL) and elevated parathyroid hormone (PTH 134 pg/mL), suggesting secondary hyperparathyroidism due to vitamin D deficiency. Arterial blood gas analysis showed a normal pH (7.35) with low bicarbonate, further confirming non-anion gap acidosis.
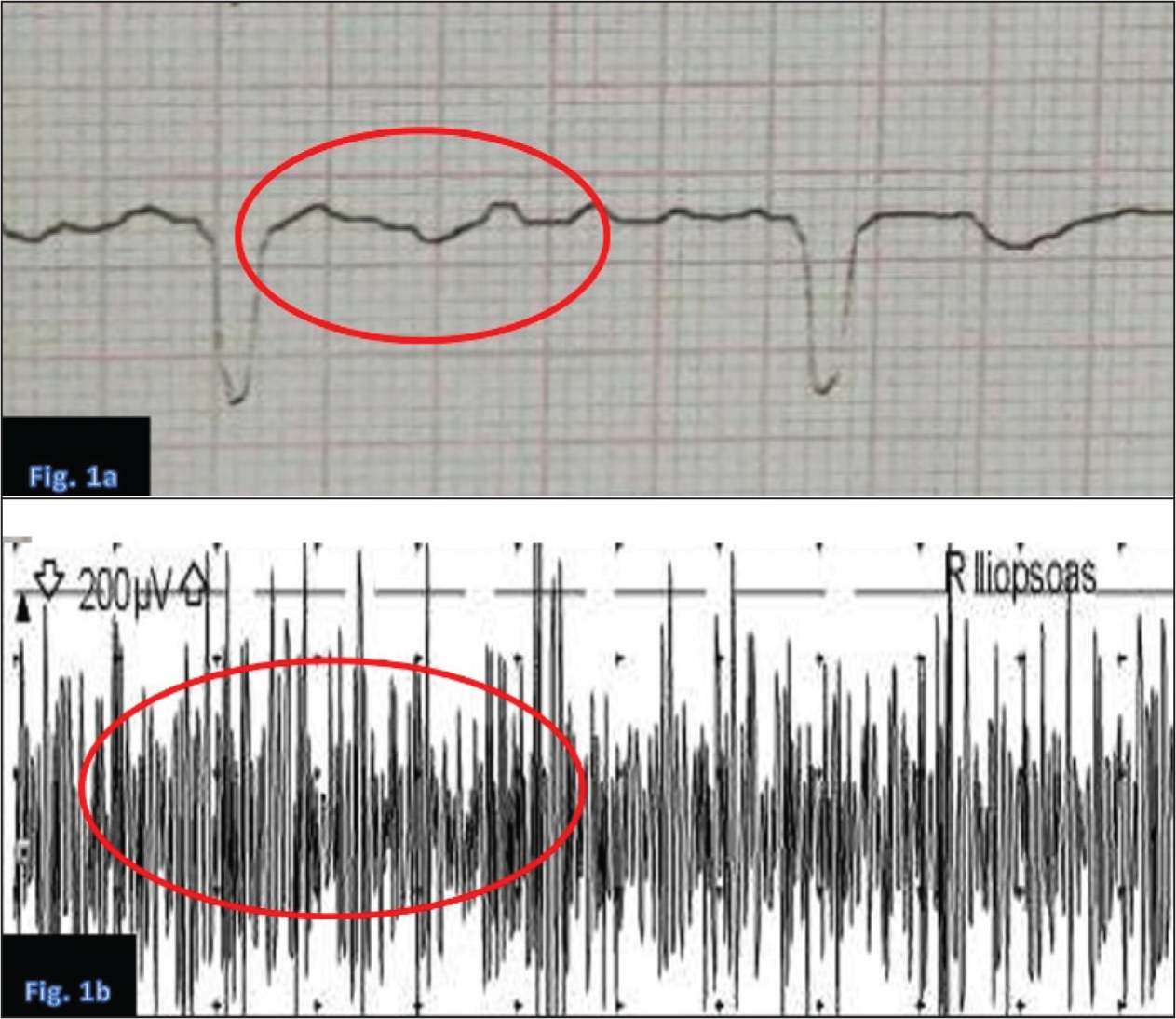
Urinalysis revealed phosphaturia, proteinuria and glucosuria, with a normal urine pH of 6.5, supporting a diagnosis of proximal RTA (type 2). The very young age of the patient and the exclusive proximal tubular involvement, despite preserved distal acidification, made the diagnosis particularly challenging and atypical for Sjögren’s syndrome. Autoimmune workup revealed positive anti-SSA/Ro antibodies (titer, 100 AU/mL), while protein electrophoresis demonstrated polyclonal hypergammaglobulinemia. These findings suggest an autoimmune basis for Fanconi syndrome, most likely associated with an early or renal-predominant variant of Sjögren’s syndrome, despite the absence of sicca symptoms or neurological involvement. An electrocardiogram (ECG) showed changes typical of hypokalaemia, including flattened T waves, U waves and prolonged QT (ventricular depolarisation–repolarisation) interval [Figure 1a]. Thyroid function tests (TSH and free T4) were within normal limits, ruling out thyrotoxic periodic paralysis (TPP). Electromyography (EMG) revealed a myopathic pattern, indicating primary muscle involvement rather than neuropathic or junctional pathology [Figure 1b]. The biochemical and serologic findings are summarised in [Table 1], highlighting the abnormalities characteristic of Fanconi syndrome. A chronological summary of the patient’s clinical course, investigations and treatment response is provided in [Table 2].
(a) Electrocardiogram showing classic features of severe hypokalaemia: Flattened T waves, U waves and prolonged QT interval. (b) Electromyography showing low-amplitude, short-duration motor unit potentials, consistent with a myopathic pattern due to electrolyte imbalance and hypophosphatemia
Laboratory investigations and interpretation in Fanconi syndrome
Clinical timeline of events, investigations and management
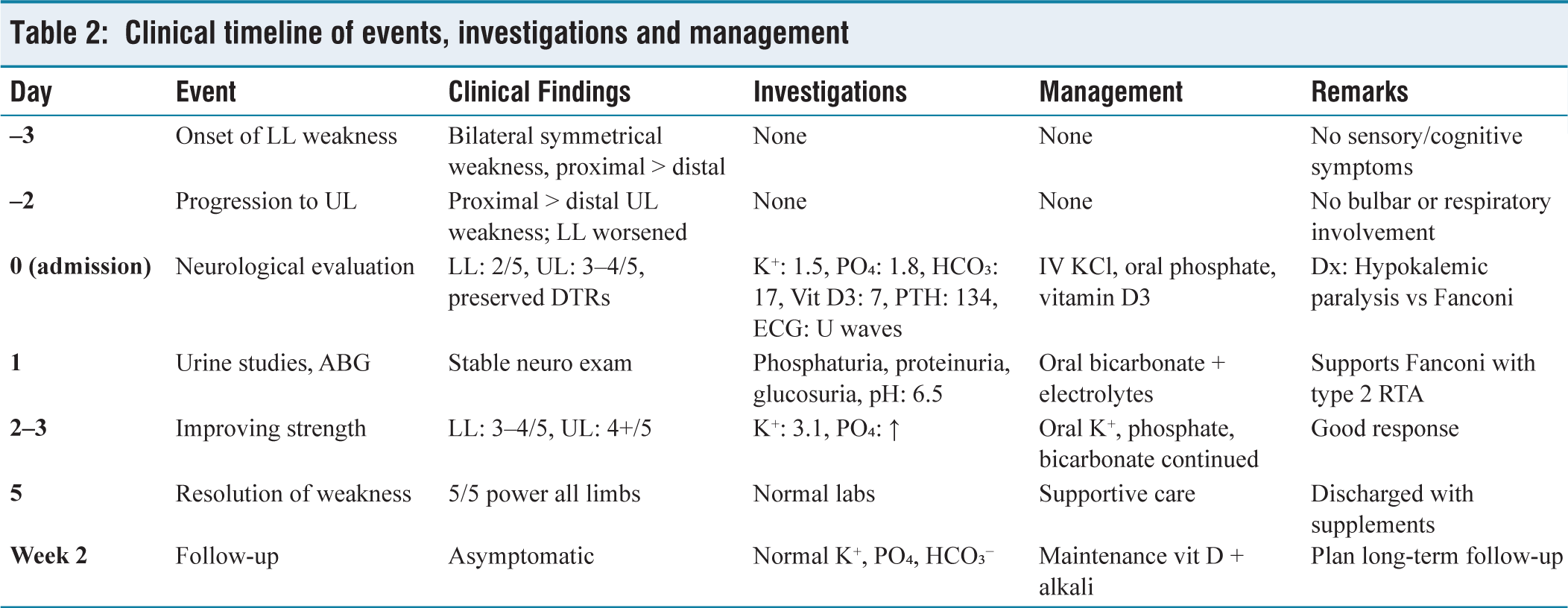
The patient was managed with intravenous and oral potassium supplementation, along with oral phosphate, vitamin D3 and sodium bicarbonate to correct the associated metabolic disturbances. She showed steady clinical improvement, with motor strength returning to near normal by day five of hospitalisation. At a two-week follow-up, the patient was asymptomatic and her serum potassium, phosphate and bicarbonate levels had normalised
Discussion
Hypokalemic paralysis should always prompt a structured diagnostic approach that distinguishes between primary and secondary causes. The hallmark of primary HypoKPP is episodic weakness with low urinary potassium excretion and normal acid-base status.[3] In contrast, secondary causes, such as TPP, RTA and Fanconi syndrome, show characteristic biochemical disturbances [Table 3].[1,5]
Differential diagnosis of hypokalemic paralysis: Clinical and laboratory comparison
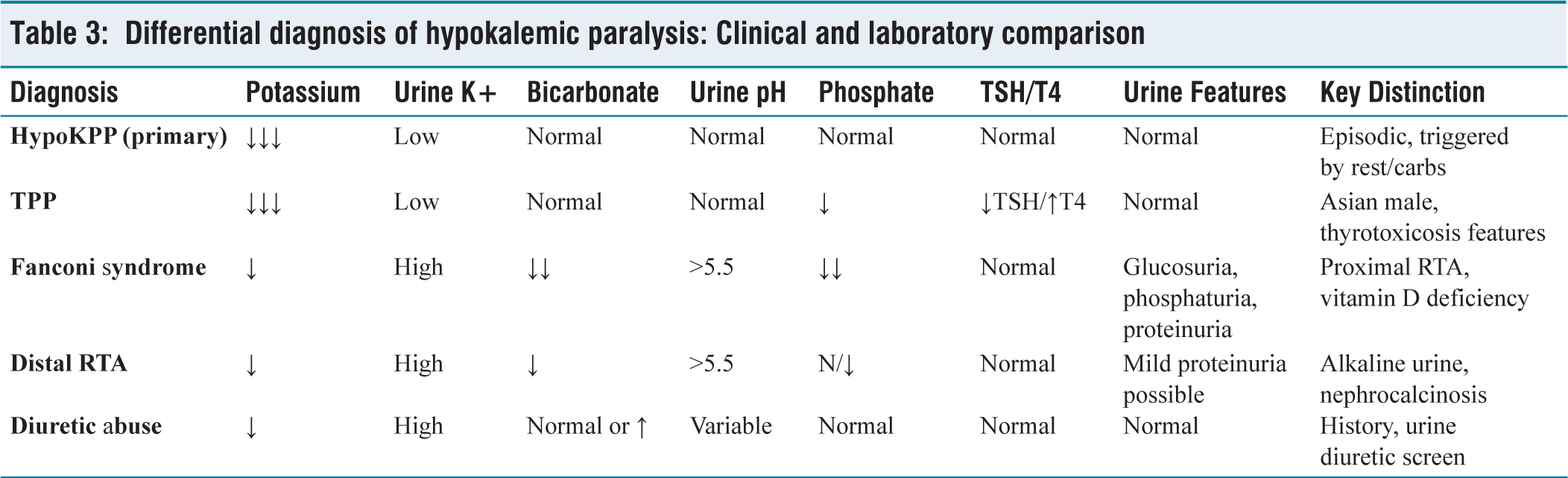
In our patient, the combination of severe hypokalaemia, normal anion gap metabolic acidosis, hypophosphatemia, glucosuria and elevated PTH indicated a proximal tubular defect. This profile is classic for Fanconi syndrome with type 2 (proximal) RTA, where bicarbonate wasting leads to systemic acidosis but preserved distal urine acidification.[6]
The electrolyte losses in Fanconi syndrome are multifactorial. Potassium is lost due to impaired proximal reabsorption and increased distal sodium delivery. Phosphate loss contributes to impaired Adenosine triphosphate (ATP) synthesis, leading to hypophosphatemic myopathy, while vitamin D deficiency accelerates muscle fatigue and bone demineralisation.[7–9] The clinical consequences of proximal tubule dysfunction, including phosphaturia, glucosuria, bicarbonaturia and hypokalaemia, Fanconi syndrome and its systemic manifestations.
The patient’s serologic profile, including elevated anti-SSA/Ro and hypergammaglobulinemia, supports an autoimmune aetiology. Sjögren’s syndrome is the most likely diagnosis, known to cause proximal RTA and Fanconi syndrome through lymphocytic tubulointerstitial infiltration. While classic sicca symptoms were absent, renal-limited or incomplete Sjögren’s syndrome has been described, especially in younger patients.[14-16]
Muscle Weakness in this Case is Multifactorial
Hypokalaemia reduces resting membrane potential, impairing depolarisation.[10]
Hypophosphatemia causes energy failure and rhabdomyolysis.[11]
Vitamin D deficiency causes secondary hyperparathyroidism, reduced calcium-phosphate reabsorption and osteomalacia.[12]
The myopathic pattern on EMG supports metabolic myopathy rather than a neuropathic process such as Guillain-Barré Syndrome (GBS). The absence of sensory signs, reflex loss and cranial involvement makes GBS unlikely.
Therapeutically, correction of electrolytes (K⁺, PO₄³⁻) and alkali therapy (HCO₃⁻) reversed the metabolic milieu. Vitamin D repletion helped in re-establishing bone-muscle-phosphate homeostasis. The rapid response with full recovery further supported the reversible metabolic aetiology.[13]
Why this Case is Unique
Atypical Age
Fanconi syndrome secondary to Sjögren’s syndrome is typically reported in adults between the fourth and sixth decades and renal involvement in younger patients is extremely uncommon. Most published cohorts describe RTA manifestations in middle-aged women, with paediatric or young adult presentations reported only sporadically.[15,16] Therefore, presentation in a 19-year-old young female is exceptionally rare and significantly expands the known age spectrum for autoimmune proximal tubulopathy.
Proximal Rather Than Distal Tubulopathy
Renal-limited Sjögren’s syndrome almost always manifests as distal RTA (type 1), secondary to impaired distal acid secretion and intercalated cell dysfunction.[5,14] In contrast, proximal tubular involvement leading to full-blown Fanconi syndrome, with phosphaturia, glucosuria and bicarbonaturia, is distinctly uncommon and reported only in isolated case reports and small series.[14,16] The presence of proximal RTA with a complete Fanconi phenotype in this patient therefore represents a highly atypical renal manifestation.
Renal-limited Autoimmune Presentation
The patient demonstrated no sicca symptoms and no extrarenal systemic involvement, which normally raises diagnostic suspicion for Sjögren’s syndrome. Despite this silent clinical phenotype, she exhibited high-titer anti-SSA/Ro antibodies and polyclonal hypergammaglobulinemia, reflecting active autoimmunity. Renal-limited or early Sjögren’s variants are increasingly recognised, with tubulointerstitial nephritis preceding glandular symptoms in a subset of patients, particularly younger individuals.[15,16] This combination, strong serologic autoimmunity without classical glandular features, supports a renal-predominant phenotype of Sjögren’s syndrome.
Neuromuscular-predominant Presentation
The dominant manifestation in this case was acute flaccid quadriparesis, rather than classic renal or systemic symptoms. The combination of severe hypokalaemia, hypophosphatemia and a myopathic pattern on EMG is consistent with metabolic myopathy caused by electrolyte depletion and proximal tubular dysfunction.[10-12] This presentation highlights how neuromuscular symptoms may overshadow underlying autoimmune renal pathology, further complicating timely diagnosis.
Clinical Implications
This case illustrates several important clinical lessons. Renal-limited Sjögren’s syndrome can present without sicca symptoms or systemic features and may manifest solely as proximal tubular dysfunction leading to Fanconi syndrome, a pattern described only in isolated cases.[14-16] In young patients with unexplained hypokalemic paralysis, hypophosphatemia or non-anion gap metabolic acidosis, clinicians should maintain a high index of suspicion for autoimmune tubulointerstitial disease, even when classic glandular symptoms are absent.[15,16] Early recognition allows timely correction of electrolyte disturbances, prevents recurrent episodes of paralysis and may reduce long-term renal morbidity. Incorporating autoimmune serology, particularly anti-SSA/Ro antibody testing, into the evaluation of atypical proximal RTA can significantly expedite diagnosis and improve clinical outcomes by identifying renal-predominant Sjögren’s syndrome at an early stage.[14-16] A stepwise approach to differentiate primary from secondary hypokalemic paralysis is outlined in [Figure 2].
Diagnostic algorithm for evaluating acute hypokalemic paralysis. The pathway highlights how metabolic and renal investigations can help differentiate primary periodic paralyses (HypoKPP, TPP) from secondary causes such as Fanconi syndrome. High urinary potassium with metabolic acidosis and proximal tubule solute wasting is diagnostic of Fanconi syndrome with type 2 RTA
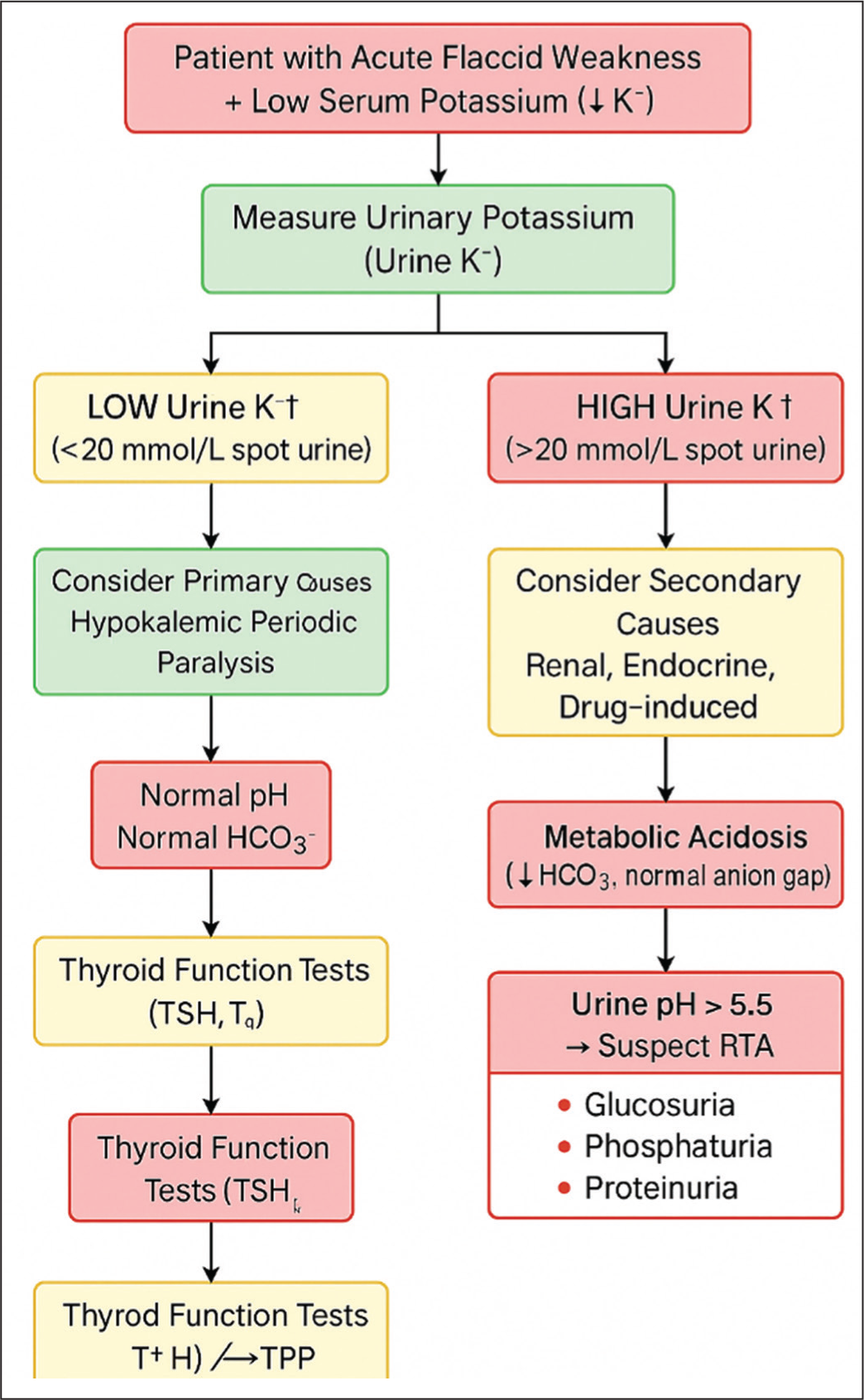
Conclusion
This case underscores the importance of evaluating autoimmune causes in unexplained proximal RTA and Fanconi syndrome. Even in the absence of sicca or neurological symptoms, anti-SSA/Ro positivity and hypergammaglobulinemia may indicate an early or renal-predominant form of Sjögren’s syndrome, guiding appropriate management and long-term follow-up. The combination of proximal tubulopathy, an uncommon renal manifestation of Sjögren’s and the unusually young age at presentation makes this an instructive case, emphasising the need for heightened clinical suspicion and early detection of renal-limited autoimmune disease.
Learning Point
Autoimmune Fanconi syndrome may present with isolated renal manifestations such as proximal RTA and hypokalemic paralysis, even in the absence of classical systemic features of Sjögren’s syndrome.
Footnotes
Acknowledgements
The authors would like to thank the Departments of Neurology, Internal Medicine and Nephrology at Indraprastha Apollo Hospitals for their collaborative support in the diagnosis and management of this case.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Funding
The authors received no financial support for the research, authorship and/or publication of this article.
Institutional ethical committee approval number
Not Applicable (NA).
Informed consent
Written informed consent was obtained from the patient for publication of this case report and accompanying images.
Credit author statement
Dr Ramesh Krishnan: Conceptualisation, clinical management and manuscript drafting.
Dr Pushpendra Nath Renjen: Neurological oversight, supervision and critical review.
Dr Avinash Gosawmi: Clinical evaluation, literature review and manuscript editing.
Data availability
All relevant data supporting the findings of this case report are available from the corresponding author upon reasonable request.
Use of artificial intelligence
No AI tools were used in the preparation or writing of this manuscript.