
Abstract
Background:
Hearing impairment is the most prevalent sensory deficiency among humans. Scientists have been actively studying the genetic factors behind human hearing loss in recent years. A considerable proportion of hearing impairment cases can be due to genetic aetiologies. Numerous specific genes have been linked to inherited hearing impairments. At present, scientists globally are engaged in genomics-driven research and development related to hearing.
Methods:
The search for recent research articles on gene therapy for hearing loss was done with databases Scopus, PubMed, Medline and Google Scholar. This review article discusses the gene therapy for hearing loss.
Results:
Screening approaches, coupled with next-generation sequencing technologies, are being employed to analyse groups of individuals affected by hearing loss who are suspected to share common genetic mutations. This approach aims to accurately diagnose patients with hearing impairments.
Conclusion:
This review article discusses gene therapy techniques for the restoration or preservation of hearing, focusing on methods for delivering genes to target cells using viral or non-viral vectors. The review also covers technology targeted at creating gene therapy as a potential therapeutic option in the near future and emphasises recent developments in gene therapy for hearing loss.
Introduction
The sense of hearing is essential to many aspects of daily life, including the enjoyment of music, the ability to locate sounds in space and interpersonal communication.[1 Hearing impairment in children hampers the development of speech and ability to read results in poor academic performance and ultimately affects employability.[2 If the hearing impairment is untreated, it may manifest cognitive decline and dementia.[3 Early diagnosis and intervention are helpful for solving such disabilities. Universal newborn hearing screening is now widely implemented for getting early diagnosis and intervention.[4 Gene therapy has advanced significantly in the last several years as a means of treating hearing loss, especially sensorineural hearing loss (SNHL) in vivo in animal models. However, a number of obstacles need to be removed before these technologies are used in clinical settings. These challenges include getting across the blood–labyrinth barrier, improving delivery vehicle specificity and efficacy, refining surgical access methods and validating novel treatment targets.
Methods of literature search
Finding current research articles on gene therapy for hearing loss requires the application of several methodical techniques. Prominent online databases like Scopus, PubMed, Medline, and Google Scholar were searched. PRISMA (Preferred Reporting Items for Systematic Reviews and Meta-Analyses) standards were followed in the development of a search strategy. This approach involved manual citation searches to find more pertinent research and concentrated on the abstracts of published articles. Eligibility criteria encompassed randomised controlled trials, observational studies, comparative studies, case series and case reports. These efforts aimed to comprehensively gather and assess current literature on gene therapy’s efficacy in treating hearing loss. This study includes a total of 72 articles categorised as follows: 15 case reports, 22 case series and 35 original articles [Figure 1]. It specifically focuses on gene therapy for hearing loss, covering various aspects such as the epidemiology and causes of hearing loss, genes associated with hearing loss, different approaches to gene therapy, methods used in gene therapy and surgical procedures related to gene therapy. The analysis aims to encourage further research into gene therapy for sensorineural hearing types of hearing loss, with a goal of advancing techniques for more effective management of hearing loss through advanced gene therapy methods.
Methods of literature search

Epidemiology
As of right now, hearing loss is the most common sensory impairment in humans and one of the most common chronic illnesses affecting the elderly. The World Health Organisation estimates that 432 million adults and 34 million children worldwide suffer from hearing impairments. By 2050, estimates suggest that this figure might climb to 700 million, or about 10% of the world’s population.[5 As individuals age, the prevalence of hearing impairment increases significantly, affecting about 60% of adults over the age of 50.[6 This demographic shift highlights the pressing need for effective solutions, given that hearing loss contributes to an annual global economic burden of approximately US $750 billion.[7 Genetic hearing impairment affects approximately 1 in 500 newborn babies and is implicated in 50%-60% of all cases of deafness over a lifetime. Developed nations are presenting higher percentages of genetic hearing impairment compared to other regions.[8
Human inner ear
The inner ear of the human being is a compact, fluid-filled sac-like structure surrounded by the hardest/densest bone in the body. This intricate three-dimensional bony framework resides at base of the skull. Sound energy is transmitted to the cochlear fluid through vibrations of tympanic membrane and the ossicles in the middle ear, generating a travelling wave along the basilar membrane. The cochlea’s length and the stiffness of the basilar membrane allow for the differentiation of sound frequencies.[9 As a result, these vibrations activate mechanotransduction in hair cells, specialised sensory cells seen in an organ of Corti. Hair cells transform mechanical stimulation into electrical depolarisation. These electrical signals generated by the inner hair cells are transmitted through the spiral ganglion and eventually processed in the auditory cortex of the temporal lobe.[10 To attain sufficient viral titres for gene therapy, it is essential to directly inject viral vectors into the inner ear. Commonly used injection techniques are through the round window membrane, canalostomy, cochleostomy into either the endolymph or perilymph and a combination of the round window membrane approach with canal fenestration. These methods enable precise delivery of viral vectors to the inner ear for therapeutic applications.[11
Hearing loss
Three categories of hearing loss are commonly used to describe hearing loss: mixed, sensorineural and conductive. A problem in the outer ear, tympanic membrane (eardrum) or ossicular chain of the middle ear that prevents sound waves from reaching the cochlea is known as conductive hearing loss.[12 Damage to the cochlea’s sensory hair cells that convert sound waves into electrical impulses or to the afferent nerves that carry this information to the brain causes SNHL. Ototoxic medication usage, loud noise exposure and ageing are all potential causes of this kind of hearing loss. Additionally, hereditary factors play a crucial role, where genetic mutations in hearing-related genes or genetic predisposition can increase susceptibility to cochlear damage and age-related deterioration. Rapid advances in genomics have made it possible to more effectively connect genes linked to hearing loss through the use of high-resolution genetic and physical maps, genomic and complementary deoxyribonucleic acid (cDNA) libraries. The creation of a human embryonic cochlear cDNA library has been especially helpful in the process of cloning several genes linked to hearing impairment. This approach aids in identifying and understanding the genetic basis of various forms of hearing loss.[13
Aetiology of hearing loss
The aetiologies of hearing impairment can vary widely, but genetic mutations are responsible for approximately 70%-75% of cases of congenital deafness.[14 In contrast, the genetic factors contributing to late-onset hearing loss are less understood, as this type of hearing impairment is often affected by environmental factors like noise exposure, medications and infections. Understanding these complex interactions between genetics and environment is crucial for developing effective strategies for the prevention and treatment of hearing loss across different age groups.[15 Studies conducted on relatives have revealed that a genetic predisposition is a major risk factor for age-related hearing loss.[16 While deafness can impact many cell types in the auditory pathway, one of the most prevalent forms of hearing loss is caused by impairment or loss of the auditory nerve fibres that make synaptic connections in the cochlea and the sensory hair cells. SNHL is the term for this kind of hearing loss.[17 In otherwise healthy newborn babies with hearing impairment not attributed to noise exposure, ageing or trauma, the cause is typically genetic or, in some cases, infectious due to prenatal cytomegalovirus infection. These factors represent the primary aetiologies of hearing impairment in such cases.
Hearing loss and genes
Approximately 50%-60% of the children born with hearing loss are associated with genetic abnormalities.[18 There is a total of 112 non-syndromic hearing loss genes that have been found: 71 autosomal recessive, 45 autosomal dominant, 5 X-linked and one non-syndromic gene.[19 In most populations, the most common cause of severe-to-profound non-syndromic hearing loss is the autosomal recessive mutation of the gap junction protein beta 2 (GJB2) gene. Connexin 26 mutations are the most common cause of genetic deafness in humans. Connexin 26 is encoded by GJB2 gene. Conversely, mild-to-moderate hearing loss is frequently attributed to autosomal recessive mutations in the STRC gene.[20 It is noteworthy that approximately 30% of inherited hearing loss cases are associated with a syndrome, indicating a syndromic component in a significant proportion of cases involving genetic hearing impairments.[21 Syndromic deafness tends to exhibit less genetic heterogeneity compared to non-syndromic forms, but it can still involve multiple loci. In several syndromes, more than one genetic locus has been identified, indicating that the genetic basis of syndromic hearing loss can be complex and involve contributions from multiple genes or genetic regions.[22 Currently, there are 11 syndromes associated with hearing loss, collectively involving 47 genes associated with syndromic hearing loss. There are different inheritance patterns for these genes: four can be inherited in both autosomal dominant and recessive patterns, two are X-linked recessive and 27 are autosomal recessive. This diversity in genetic inheritance underscores the complexity and variability of syndromic forms of hearing loss.[23 Usher syndrome has been linked to mutations in the Usher syndrome 1C gene, which codes for the protein harmonin. A basic helix loop helix transcription factor required for hair cell development is encoded by the gene atonal homolog 1 (ATOH1). With the use of an adenovirus vector and the cochleostomy technique, the ATOH1 can be introduced into the afflicted cochlea.
Treatment
Treating hearing loss poses significant challenges for clinicians. Conductive hearing loss, which involves issues in outer or middle ear, can often be corrected surgically in many patients. However, SNHL, which typically involves damage to the inner ear or auditory nerve pathways, is usually irreversible and leads to permanent hearing impairment. Management strategies for SNHL typically focus on hearing aids or cochlear implants to improve hearing function, as medical or surgical interventions to restore hearing are limited.[24 Hearing rehabilitation is achievable through the use of hearing devices, which can either be worn externally (like hearing aids) or implanted internally (like cochlear implants). Despite significant advancements in both hearing aids and cochlear implant technologies, the quality of sound provided by these devices does not match that of a normal ear. Challenges such as impaired speech perception in noisy environments and limitations in musical sound perception remain significant hurdles for cochlear implants in particular.[25 Currently, the primary treatment options for SNHL are limited to hearing aids or cochlear implants. While these devices can improve hearing, they do not fully restore natural sound perception, especially in challenging environments like noisy settings, leading to difficulties in speech recognition. Recent advancements in understanding genetic abnormalities related to deafness, coupled with developments in genetic editing and correction techniques, have sparked optimism for gene-based therapies. Gene therapy has emerged as a promising approach to address the underlying causes of hearing loss, potentially offering a more effective and targeted treatment in the future.
Gene therapy
Gene therapy primarily aims to replace, suppress or edit faulty genes to treat diseases, including those affecting the cochlea. Because of its physical isolation and the blood–labyrinth barrier, which effectively isolates it from systemic blood circulation, the cochlea is a perfect organ for these therapeutic techniques.[26 This barrier helps in delivering therapeutic agents directly to the cochlea without significant systemic effects, making it a suitable target for gene-based treatments aimed at addressing hearing loss and related conditions. The unique anatomical characteristics of the cochlea make it an ideal organ for gene therapy by minimising the risk of potential off-target effects in systemic circulation. Therapeutic vectors can be delivered directly into the cochlear fluid space, allowing for even distribution along the entire length of the cochlea and effective targeting of specific cell types. Among the various gene-based approaches, replacing a faulty gene with a normal copy has been the most extensively utilised method for treating monogenic deafness. This approach aims to restore normal gene function and address the underlying genetic cause of hearing loss, offering promising prospects for therapeutic intervention in auditory disorders.[27 The majority of studies demonstrating successful recovery in mice afflicted with hereditary deafness have focused on genes that play critical roles in the development and/or function of the postnatal mouse cochlea. These studies typically involve interventions aimed at restoring or enhancing cochlear function through gene therapy approaches that target specific genetic defects associated with hearing loss.[28,29] A study demonstrated that the efficiency of gene replacement using adeno-associated virus (AAV) vectors in the adult cochlea is lower compared to early postnatal stages. This finding suggests that there may be age-related differences in the transduction efficiency of AAV vectors in the cochlea, potentially impacting the effectiveness of gene therapy approaches for treating hearing loss in adult individuals compared to younger stages of development.[30
Goal of gene therapy
The primary goal of gene therapy in the context of treating hearing loss is to preserve or restore hearing function. Achieving this goal necessitates a comprehensive understanding of both the normal development and the abnormalities associated with the inner ear. Researchers aim to leverage this knowledge to develop innovative and effective treatments specifically targeted towards patients with genetic forms of hearing loss. By addressing the underlying genetic causes of deafness, gene therapy holds promise as a potential breakthrough in improving auditory function and quality of life for affected individuals.[31
Methods of gene therapy
Gene replacement
Gene replacement therapy aims to identify and substitute a defective gene with a normal or wild-type copy, offering a direct approach to gene therapy. This technique has demonstrated potential in treating genetic disorders such as haemophilia and Leber’s congenital amaurosis, where specific mutations in genes are known to cause disease. By restoring the function of the affected gene, gene replacement therapy aims to alleviate or eliminate the symptoms associated with these conditions, providing potential therapeutic benefits to affected individuals.[32 Studies have been conducted using murine models of Usher syndrome, Jervell and Lange-Nielsen syndrome, as well as a type of deafness caused by a deficiency in vesicular glutamate transporter-3 (VGLUT3).[33 In the first effective inner ear gene therapy trial, mice with homozygous or targeted VGLUT3 deletion were used. This research utilised adeno-associated virus serotype 1 (AAV1) to facilitate the overexpression of VGLUT3 in inner hair cells. The outcomes demonstrated sustained recovery of hearing function, partial restoration of ribbon synapse morphology and improvement in startle responses among the treated mice. This study underscored the potential efficacy of gene therapy employing AAV1 vectors to target and potentially correct specific genetic deficiencies linked to hearing loss in animal models.
Gene suppression
The purpose of chemically altered nucleic acid sequences known as antisense oligonucleotides (ASOs) is to use Watson–Crick base pairing to bind complementary ribonucleic acid (RNA) sequences. ASOs can achieve knockdown of gene expression through several mechanisms: RNase H-mediated cleavage: ASOs can hybridise to the target RNA, forming an RNA-DNA duplex. This duplex structure can then be recognised and cleaved by the enzyme ribonuclease H (RNase H), resulting in degradation of the RNA strand. Splice modulation: ASOs can interfere with alternative splicing by targeting splice sites, exons or introns in pre-mRNA molecules. This interference can lead to the exclusion or inclusion of specific exons in the mature messenger RNA (mRNA), altering the resulting protein product. These mechanisms make ASOs a versatile tool for manipulating gene expression and potentially treating genetic disorders by targeting specific RNA molecules.[34 The US Food and Drug Administration (FDA) has approved five ASOs thus far, and more clinical trials are still in progress. Patients with acquired immune deficiency syndrome (AIDS) are treated for cytomegalovirus-induced retinitis with formivirsen, the first licensed ASO.[35
Gene silencing using RNA interference
Gene silencing can occur through various mechanisms during different stages of gene expression. At the transcriptional level, gene silencing prevents the synthesis of mRNA, effectively reducing the expression of a particular gene. This method can be employed to suppress dominant mutations in heterozygous animals, where the mutant allele is inhibited or regulated negatively, enabling the wild-type allele to predominate and counteract the mutation’s effects. Alternatively, gene silencing can occur at the post-transcriptional level through RNA interference (RNAi). RNAi mechanisms involve small RNA molecules that bind to mRNA and inhibit its translation into protein. This approach effectively decreases the production of abnormal proteins encoded by mutant alleles, providing another strategy to alleviate the effects of genetic mutations linked to conditions such as hearing loss and other disorders.[36 RNA interference (RNAi) predominantly employs two types of small complementary RNA molecules: microRNA (miRNA) and small interfering RNA (siRNA). In studies involving mice exposed to excessive sound, siRNA has demonstrated effective silencing of adenosine monophosphate-activated protein kinase expression. This protein is associated with hair cell loss and cochlear synaptopathy. One major advantage of RNAi technology is its sequence specificity. This specificity enables siRNA to target and silence specific genes or mutations without impacting wild-type sequences or causing off-target effects.[37 This precision makes RNAi particularly effective for silencing dominant mutations linked to genetic disorders, such as those contributing to hearing loss. It allows for the suppression of abnormal gene activity while preserving normal gene function, thereby minimising unintended consequences.[38
Gene editing
Another method for gene therapy involves modifying genome sequences using the Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)/Cas9 system. This gene-editing technique originates from prokaryotic immune systems, which evolved to defend against phages and plasmids.[39 This is the most recent and sophisticated programmable nuclease designed specifically for genome engineering, allowing for accurate editing of the inner ear’s genome sequences.[40 Nuclease-based enzymes in genome engineering are valuable for targeting specific genome sequences and facilitating the introduction of single or double-strand DNA, which triggers the innate DNA repair mechanisms. Among these, CRISPR/Cas is widely utilised due to its versatility and ease of use across various applications. However, Cas9 requires a protospacer adjacent motif (PAM) directly following the DNA target sequence for specificity, which also restricts its clinical applicability.[41 Currently, significant efforts are focused on designing CRISPR nucleases with modified PAM specificities and reduced off-target effects, aiming to broaden their applications even further.[42
Viral vectors
The workhorse of cochlear gene therapy is viral vectors. Several research has employed lentivirus, adenovirus and AAV, helper-dependent lentivirus, herpes simplex virus, vaccinia virus, Sendai virus and vaccinia virus as viral vectors in gene therapy.[43 Out of all these viruses, AAV has emerged as the most attractive virus for cochlear gene delivery. AAV is a small virus of the family Parvoviridae and genus Dependovirus that lacks pathogenicity with minimal immunogenicity.[44 This transduces both non-dividing and dividing cells to give stable long-term gene expression by persisting as an episome in the absence of chromosomal integration.[44 The disadvantage of AAV is its very low viral capacity which is again halved when using a self-complementary AAV.[44 Inside the inner ear, AAVs show broad tropism, stable gene expression and little to no ototoxicity.[45
Non-viral vectors
Liposomes, polymeric nanoparticles and synthetic peptides are examples of non-viral vectors that offer a potent substitute method for delivering medications to the cochlea. Nanomaterials have a number of unique benefits over viruses. These are designed to exhibit low immunogenicity and toxicity, precisely target a particular cell subpopulation and multiplex in a high-throughput manner to address several gene targets and pathways at once. Recent years have seen the development of nanomaterial-based cochlear delivery systems that use liposomes, peptides or polymers.
Surgery for gene therapy
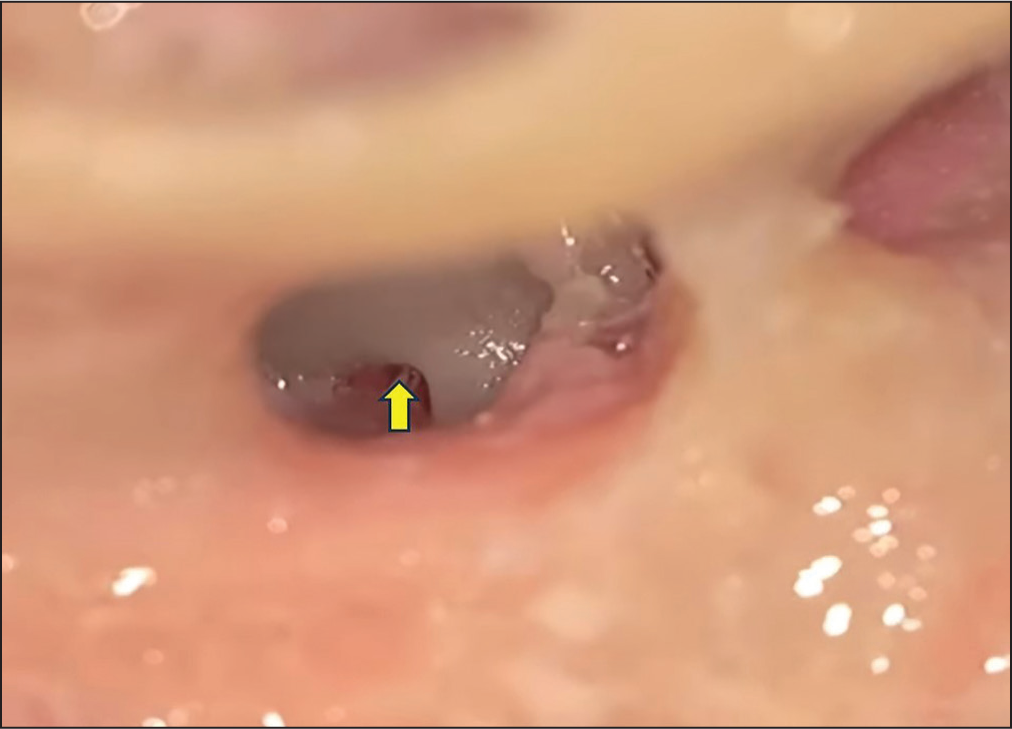
There are two important approaches used clinically such as intra- or transtympanic delivery for getting surgical access to inner ear. In these methods, a semicircular canal, an oval window or a round window membrane is used to inject the therapy into the middle ear space and allow it to diffuse into the inner ear (intracochlear). The strategy should try to maintain the patient’s remaining hearing to the greatest extent feasible.[43 The lateral wall of the cochlear basal turn, a semicircular canal or an injection through a round window membrane is among the surgical techniques used in animal models to deliver gene therapy locally. While these techniques have been shown to be safe, they do carry a risk of permanent SNHL and inner ear damage. Injections into the middle ear, known as intratympanic or transtympanic procedures, are often low-risk procedures. Adult patients may undergo this office-based procedure under topical anaesthetic. Direct approach for a round window membrane [Figure 2] can be achieved through a trans-canal surgical procedure or cochleostomy with the help of a microscope/otoendoscope. Cochleostomy directly delivers transgenes to the scala media which can be done via a hole drilled through the basal part of cochlea into cochlear endolymphatic space near round window.[43 The canalostomy method for gene therapy in mouse cochleae is an alternative to the round window membrane injection technique. This approach involves injecting adenovirus carrying bacterial lacZ through an opening in the posterior semicircular canal and guiding the cannula towards the crus commune.[46
Exposure of round window membrane via cochleostomy for the delivery of genes into scala tympani
Conclusion
Hearing loss is the most prevalent sensory impairment worldwide, affecting millions of people and significantly impacting their lives. Recent years have seen rapid advancements in diagnostics and therapeutic approaches for SNHL. Gene therapy represents a promising avenue in this field, focusing on strategies to replace, edit or suppress defective genes to treat SNHL effectively. Several studies have shown successful gene therapy outcomes in neonatal and adult mouse models of human deafness. An interdisciplinary approach that integrates new genomic data, advanced bioengineering tools and innovative surgical techniques is crucial for the clinical success of gene therapy for SNHL. However, while gene therapy holds promise as a treatment option for hearing loss, its application is currently constrained by potential side effects and ongoing studies are essential to ensure its safety and effectiveness before widespread clinical adoption can occur.
Footnotes
Acknowledgements
The author is thankful to all faculties of the Department of Otorhinolaryngology and Head and Neck Surgery, AIIMS, Bhubaneswar, for their valuable suggestions.
Declaration of conflicting interests
The author declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Funding
The author received no financial support for the research, authorship and/or publication of this article.
Institutional ethical committee approval number
Not applicable as this is a review article.
Informed Consent
Not applicable.
Credit author statement
Santosh Kumar Swain contributed to concept, literature search, data acquisition, manuscript preparation, manuscript editing and approval of the final version to be published.
Data availability
Data are available in a public, open-access repository.
Use of artificial intelligence
Not applicable.