
Abstract
Introduction:
Patients frequently present with renal impairment that does not fit the criteria for acute kidney injury or chronic kidney disease. This condition, known as rapidly progressive renal failure (RPRF), encompasses a diverse range of clinical syndromes characterized by the progressive renal impairment over days to weeks.
Case Report:
We present a case of a 37-year-old male with a short history of uremic symptoms and decreased urine output with no significant history or examination findings except pallor. The patient had a creatinine of 1.6 mg/dL in April 2022, rising to 4.4 mg/dL, then 11.6 mg/dL during May, 17.6 mg/dL in June, presented to us with a creatinine of 24.6 mg/dL, with normal kidney size and corticomedullary differentiation (CMD). Beta-2-microglobulin was elevated, and renal biopsy showed features suggestive of multiple myeloma (MM). Atypical appearing tubular casts showing 3+ patternless staining for kappa light chains and negativity for lambda light chains were observed. The patient was started on chemotherapy for MM, and renal function was gradually improving.
Conclusion:
Renal failure as the sole presentation of MM is rare, especially in young adults. This case emphasizes an atypical RPRF, particularly given the patient’s young age.
Introduction
Physicians in clinical practice often encounter progressive renal impairment of variable durations with an ambiguous or unknown etiology. Sometimes, these patients may have a time frame that neither fits as an acute kidney injury nor can we call them to be a chronic kidney disease. Of these, a clinically heterogeneous group of clinical syndromes sometimes presents with the progressive renal impairment over days to weeks. The initial clinical diagnosis of these cases may be called as rapidly progressive renal failure (RPRF), encompassing a myriad etiology. These patients often have a normal-sized kidney, and renal histopathology shows lesions affecting any or a combination of the three traditional renal compartments: glomerular, tubulointerstitial, or vascular.[1] RPRF can be regarded as a critical situation or a “Renal Emergency” because without timely intervention, it can lead to irreversible end-stage renal disease, requiring lifelong renal replacement therapy.
Multiple myeloma (MM) is a type of cancer that originates from plasma cells. It is characterized by the presence of a serum monoclonal spike (M-Spike) >3g/dL or more than 10% clonal plasma cells in the bone marrow. In addition, the diagnosis of MM requires the presence of at least one myeloma-defining event, which includes anemia, hypercalcemia, renal insufficiency, or bone lesions.[2] While renal impairment is a frequent presenting symptom, it rarely is the singular presenting symptom of MM and presents primarily as an RPRF without the classical symptoms occasionally. However, RPRF as a presenting complaint in the young is rare. In this case, we present a young adult patient who presented with RPRF as the only manifestation without any classical symptoms of plasma cell dyscrasia.
Case Report
A 37‑year‑old male patient, nondiabetic nonhypertensive, apparently healthy before presentation was admitted with a short 2‑week history of nausea, hiccups, decreased urine output, and generalized fatigue with weakness. He had a history of an RTA 5 months back, which was treated in a different hospital with the persistence of pain in the nape of the neck where he got injured during the accident. There was no history of fever, hemoptysis, joint pain, photosensitivity, pain in the abdomen, hematuria, pedal edema, orthopnea, nor any suggestive history of recent infection. There was no other significant past medical history. Family history and social history were noncontributory. The patient had a long‑standing history of nonsteroidal anti‑inflammatory drug use for his pain, which he often took as an over‑the‑counter drug. Physical examination of the patient was notable for the presence of pallor but was otherwise unremarkable.
Laboratory values on admission were notable for a creatinine of 24.6 mg/dL, BUN of 134 mg/dL, potassium of 5.7 mEq/L, and hemoglobin of 7.5 g/dL. Urinalysis was normal for the patient and urine sodium was 56 mEq/L, whereas liver function tests and serum calcium (9.1 mg/dL) were within normal ranges. The patient’s volume status, vital signs, and these laboratory values played a crucial role in differentiating prerenal causes from intrinsic renal causes. Renal ultrasound was performed, which showed a right kidney measuring 9.5 cm × 6.5 cm and a left kidney measuring 10.5 cm × 6.1 cm. The size of the kidneys was within the normal range, and there were no indications of mass lesions, hydronephrosis, or calcifications. The corticomedullary differentiation was also found to be normal. On revisiting the patient’s medical records, he had a creatinine of 1.6 mg/dL in April 2022, which rapidly increased to 4.4 mg/dL and then to 11.6 mg/dL in May 2022, with a last record of 17.6 mg/dL in early June 2022. He had collapsed C2 vertebrae on magnetic resonance imaging spine, which was mainly attributed to the old accident. Given the rapidly deteriorating renal status, the patient underwent further laboratory workup. The serum electrophoresis of the patient was negative, but beta‑2 microglobulin assay level was 72,800 mg/mL. The patient was given urgent hemodialysis, and a decision was taken to perform a renal biopsy for the patient.
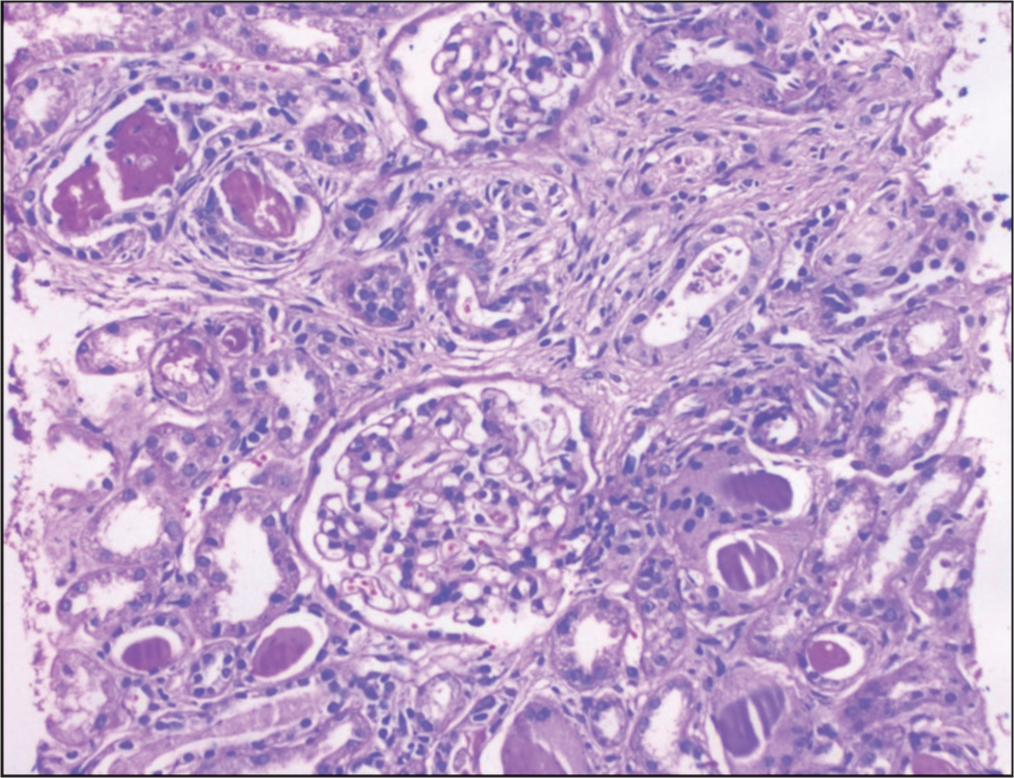
The renal biopsy clinched the diagnosis for the patient. On microscopy, glomeruli show nonproliferative morphology with no evidence of crescent formation, tuft necrosis, subendothelial/congophilic deposits, or intracapillary thrombi. Tubules showed evidence of severe injury with the inspissation of scattered hyaline, granular, and many “atypical” casts with waxy to granular appearance, brittle appearance, and “fracture” planes also associated with inflammatory and epithelial cell reactions [Figure 1].
The DIF of the renal cortical parenchyma was negative, but many atypical appearing tubular casts showing 3+ patternless staining for kappa light chains and concurrent negativity for lambda light chains were observed.
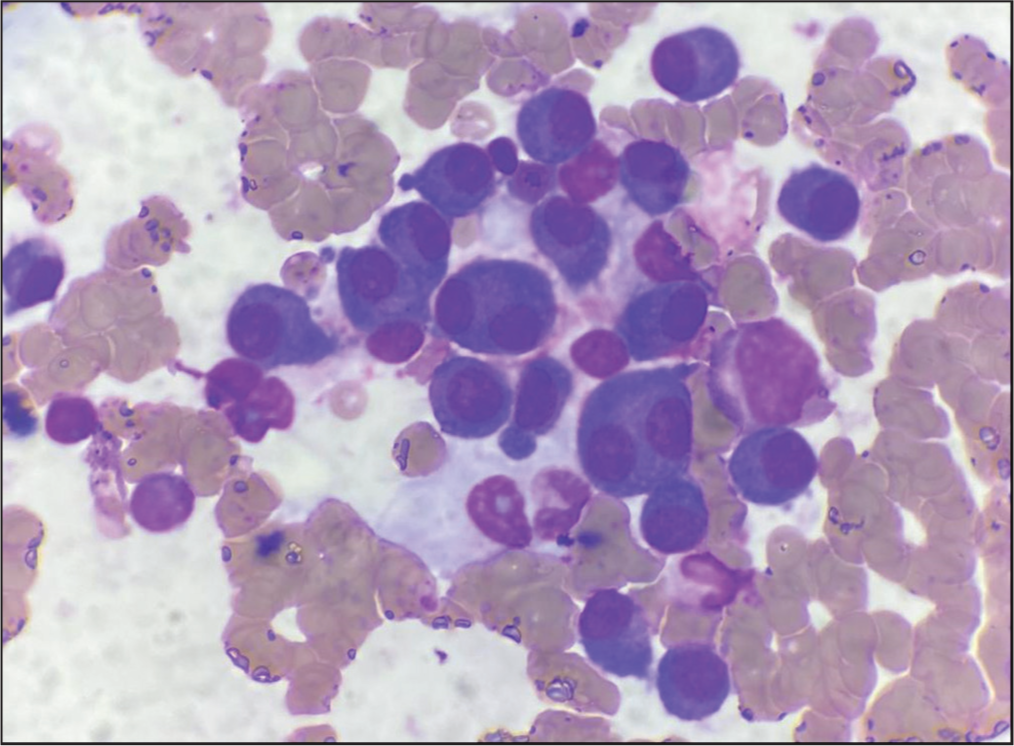
Concurrently, a bone marrow aspiration study was also done, which revealed the presence of a high percentage of plasma cells (62%) [Figure 2].
Renal biopsy (LM)
Bone marrow assay
A diagnosis of MM with cast nephropathy presenting with RPRF was thus made. The patient was started on chemotherapy for MM, and renal function was gradually improving on treatment with serum creatinine showing a declining trend (discharged with a creatinine of 4.3 mg/dL), with adequate urine output and kept on close follow-up.
Discussion
MM is characterized by the malignant growth of plasma cells. The disease is identified through a set of symptoms known as “CRAB” features, which include hypercalcemia, renal failure, anemia, and bone lesions. These symptoms are instrumental in the diagnostic evaluation of MM. Hypercalcemia and bone pain, resulting from lytic bone lesions and heightened osteoclastic activity, are observed in approximately 28%–58% of MM patients on presentation, respectively. Anemia, found in about 73% of MM patients at the time of diagnosis, is the most prevalent manifestation of the disease.[3] Renal failure is the focus of this case report and occurs due to multiple pathogenic mechanisms. On initial presentation, around 48% of individuals diagnosed with MM display elevated levels of creatinine.[3]
Renal dysfunction in MM can be a result of either a paraprotein or a nonparaprotein-associated renal complication, with tubular nephropathy often being the final pathogenic pathway in the renal injury.[4]
In a study of MM published by Prakash et al., it was found that, before diagnosis of the condition, renal dysfunction most commonly presented as acute renal failure, followed by chronic renal failure, and finally, nephrotic syndrome. This study found that 84% of the study population presented with renal impairment before a diagnosis of MM.[5] According to a study conducted at the Oxford Regional Renal Unit, which included 56 patients diagnosed with MM, it was observed that half of the patients experienced renal failure as the initial manifestation of the disease. However, a significant majority of these cases also exhibited hypercalcemia, which could have contributed to the development of renal failure.[6]
In the absence of immune complex deposition, the Medical Oncology Journal in 2011 presented a case report of a patient who exhibited renal dysfunction evident in laboratory results. The patient was discovered to have light-chain deposition and findings consistent with membranoproliferative glomerulonephritis. At the initial presentation, the patient did not display any signs or positive diagnostic tests indicating MM. However, after 28 months, the patient fulfilled the diagnostic criteria for MM. This report presented the idea that renal disease can manifest as a presenting symptom or occur before the onset of MM.[7] Another study done analyzed patients with MM who were referred only for renal failure without any features suggestive of MM. This study found that the only consistent presentation was anemia (as was in our case) and concluded that, in adults over the age of 50 years, MM should be taken into consideration as a potential underlying cause of unexplained acute renal failure.[8]
Conclusion
Our case study provides a solid base and enables us to take a step further in considering the diagnosis of MM in patients with acute or rapidly progressive renal dysfunction, even if they do not exhibit the typical features of MM. While there is ample evidence linking renal failure to MM, there is limited evidence suggesting that renal failure alone can be the primary manifestation in MM patients. Our case report demonstrates that rapid progressive renal dysfunction can be the initial and only presentation of MM, even in young adults. Renal biopsy plays a significant role in such cases, and it is important to consider the possibility of MM even if the patient’s age does not align with commonly observed trends.
Footnotes
Acknowledgements
We would like to thank Dr. Alok Sharma for his invaluable input in the histopathology of the patient.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of Patient Consent
The authors certify that all appropriate consent forms were obtained, which contained the patient’s consent for his clinical information to be reported on a scientific platform. The patient understood that his name and personal details would not be published anywhere, and due efforts would be made to conceal his identity, although anonymity cannot be guaranteed.
CRediT Author Statement
The authors mentioned in the manuscript: (1) They have made substantial contributions to the article’s concept, design, data acquisition, analysis, or interpretation. (2) They have either drafted the article or provided critical revisions that are important for its intellectual content. (3) They have given their approval for the publication of the final version. (4) They have agreed to take responsibility for the work by addressing any concerns related to its accuracy or integrity and ensuring appropriate investigation and resolution.
Data Availability
Available in records department of hospital.
Use of Artificial Intelligence
Not used during any part of the preparation of the article.