
Abstract
Introduction:
Sjogren’s syndrome (SS) is an autoimmune chronic inflammatory disorder affecting women in their fourth to sixth decade, affecting gastrointestinal, and musculoskeletal systems. A 35-year-old female with SS with polymyositis (PM) presented with symptoms of weakness in all four limbs, difficulty in sitting, rising, swallowing solid foods, vomiting, and difficulty in climbing stairs. She was diagnosed with SS in 2018 and was treated with prednisolone, Vitamin D, calcium, pyridoxine, methylcobalamin, artificial tears, pilocarpine, and painkillers. However, a muscle biopsy was never done.
Methods:
The data were collected from the patient’s file along with her consent when she came for follow-up.
Results:
A muscle biopsy was done at our center to confirm the diagnosis of PM. The patient was prescribed IV prednisolone and other symptomatic treatment until symptoms resolved and was discharged with oral drugs when they were manageable.
Conclusion:
The diagnosis of primary SS along with PM is a rare occurrence. Although it does not change the treatment plan much, its diagnosis is very important for managing any complications that may arise from it. Patients’ noncompliance and loss of follow-up can create issues in the treatment. Such cases help in forming the guidelines for the future and restructuring the classification of autoimmune conditions.
Introduction
Sjogren’s syndrome (SS) is an autoimmune chronic inflammatory disorder that usually affects women in their fourth to sixth decade. It can be primary or secondary to other autoimmune conditions like systemic lupus erythematosus and can present with sicca syndrome or involving gastrointestinal or musculoskeletal systems. Sjogren is diagnosed clinically and by testing the presence of autoantibodies such as anti- Ro/SSA or Anti-La/SSB; however, a biopsy of the salivary gland is a gold standard test.[1] Myositis and SS can be due to adverse effects of drugs, copresentation of other autoimmune conditions, or true myositis.[2] The latter is very rare, with no exact data available on its prevalence.
Case Report
A 35-year-old female who had a known case of SS with polymyositis (PM) presented to outpatient department with complaints of weakness of all four limbs, difficulty in sitting from a supine position, difficulty in rising from a squatting position, difficulty in swallowing solid foods and episodes of vomiting, difficulty in running and climbing stairs, dry mouth, and dry eyes. In elaborating on the patient’s history, it was discovered that she was diagnosed with SS in 2018. The patient’s vitals were normal, with a pulse of 70 beats/min and blood pressure of 112/76 mmHg. She was conscious, cooperative, and oriented to time, place, and person. Physical examination showed pale conjunctiva. Muscle tone and power were decreased; 4/5 in upper limbs and 3/5 in lower limbs. Reflexes were normal in all limbs.
The patient’s history involves visiting multiple hospitals before reaching the diagnosis of the current condition. In 2017, the patient went to a private hospital complaining of weakness in all limbs. Investigations were done, and the patient was given a provisional diagnosis of bilateral asymmetrical demyelinating axonal sensory > motor neuropathy affecting lower limbs > upper limbs. Further investigations were required to reach a proper diagnosis. Electromyography (EMG) studies showed decreased muscle unit action potentials. Based on this, she was diagnosed with girdle muscular dystrophy and started on oral prednisolone 10 mg daily. With little signs of improvement over the months, the patient went to another hospital with the same complaints. The patient was admitted, and investigations were carried out. Her laboratory reports are shown in Table 1.
Lab investigations from previous centre
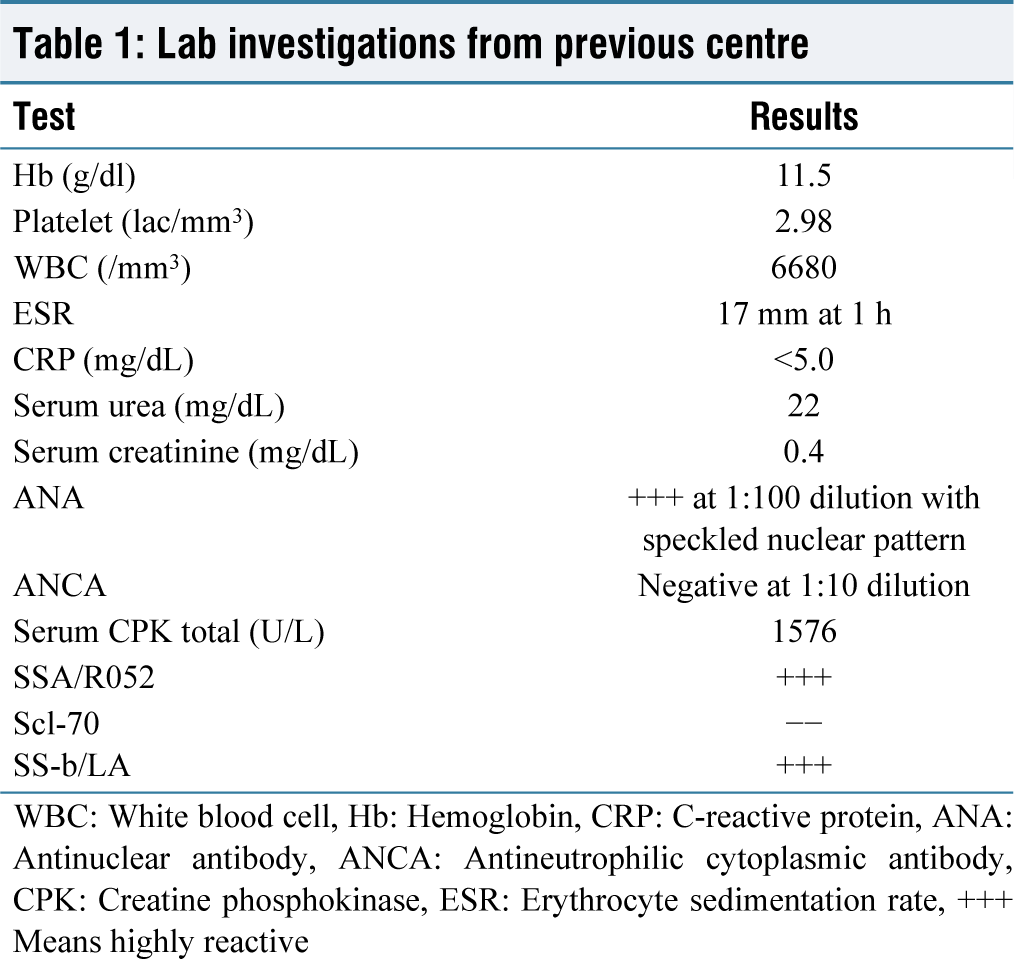
WBC: White blood cell, Hb: Hemoglobin, CRP: C-reactive protein, ANA: Antinuclear antibody, ANCA: Antineutrophilic cytoplasmic antibody, CPK: Creatine phosphokinase, ESR: Erythrocyte sedimentation rate, +++ Means highly reactive
Considering all the reports, the patient was finally diagnosed with SS with PM. Treatment was started with prednisolone 10 mg/day, Vitamin D, calcium, pyridoxine, methylcobalamin, artificial tears, pilocarpine, and painkillers as required.
The patient was admitted to our hospital, and investigations were carried out, along with examining the old reports. A Schirmer’s test was done, which was abnormal, with 4 mm in each eye. We realized that the biopsy was never done to confirm the diagnosis of PM. Laboratory reports are shown in Table 2. A muscle biopsy was taken from the right quadriceps muscle and was sent for histopathological examination. The results showed that the muscle fibers were in different necrosis, degeneration, and regeneration stages with endomysial inflammation. The histopathology section of the muscle biopsy is shown in Figure 1. Repeat immunoassays were positive for antinuclear antibody (ANA), sjogren’s syndrome -type A (SSA)/Ro52, and sjogren’s syndrome -type B (SSB)/LA. The patient fulfilled the diagnostic criteria by European alliance of associations for rheumatology (EULAR) for both SS and PM.
She was started on IV prednisolone and other symptomatic treatment until the symptoms resolved, and then she was continued on oral drugs as before. It was found that the patient had similar complaints as she was not going on regular follow-ups and was skipping medications. She was explained to comply with the medicines and follow-ups and was discharged with oral drugs when the symptoms were manageable.
Lab investigations at out centre
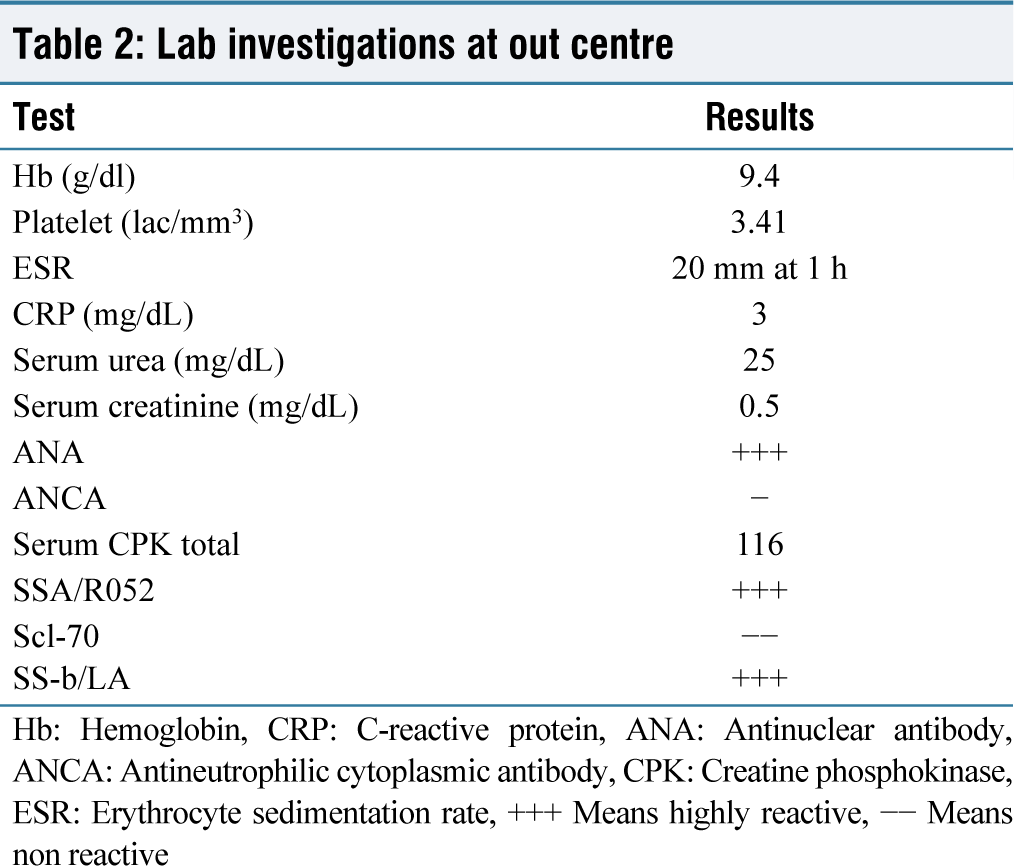
Hb: Hemoglobin, CRP: C-reactive protein, ANA: Antinuclear antibody, ANCA: Antineutrophilic cytoplasmic antibody, CPK: Creatine phosphokinase, ESR: Erythrocyte sedimentation rate, +++ Means highly reactive, −− Means non reactive
Histopathology section of muscle biopsy
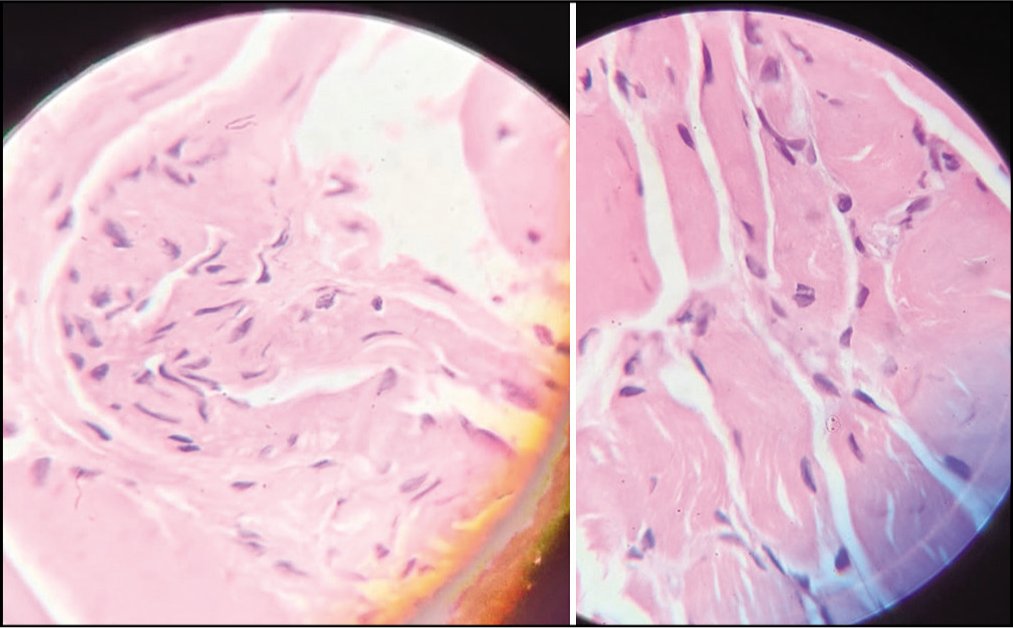
Discussion
SS is more common in females and usually occurs in the age group of 40–60 years. It appears with symptoms such as dry eyes, mouth, and vagina due to the disease activity in extra glandular glands. Sometimes Sjogren occurs due to other diseases called secondary SS.[1] PM is a type of idiopathic inflammatory myopathy that is also more common in females, unlike inclusion body myositis, which is more common in males. It presents with proximal muscle weakness. PM can occur alone or along with other autoimmune or connective tissue diseases. The pathophysiology involves the immune system acting against the endomysial and perimysial muscle fibers.
SS is usually diagnosed with clinical features and immunoassays, but the 2016 EULAR criteria are also widely accepted.[1] However, in PM, there are multiple criteria, but each has limitations, and none is widely accepted, so the diagnosis relies on clinical findings like proximal muscle weakness and investigations such as elevated muscle enzymes, EMG, and biopsy. The diagnostic classification of PM is shown in Figure 2.[3]
Diagnosis classification of polymyositis
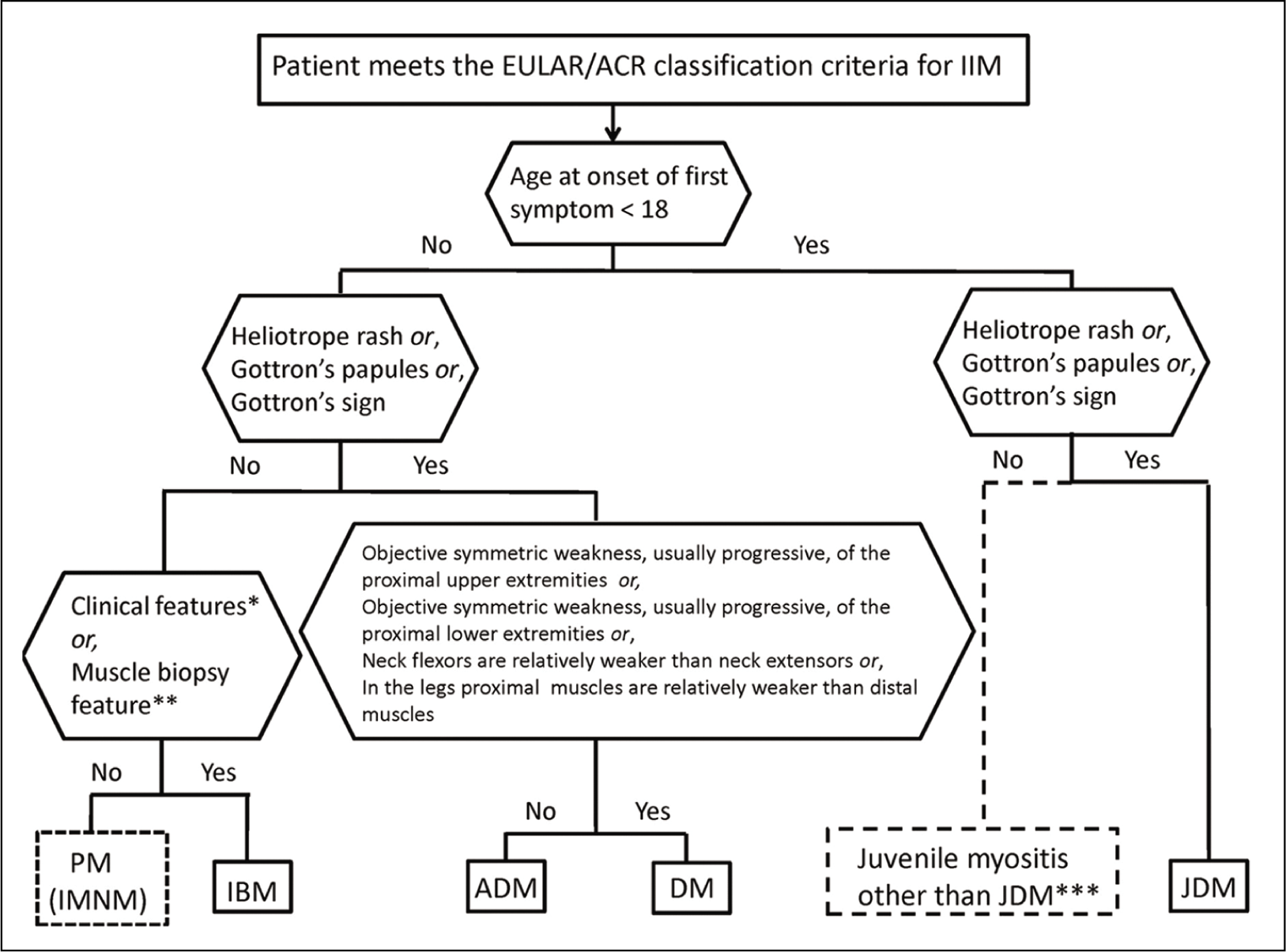
The occurrence of PM with SS is rare. At present, no studies show the incidence and prevalence of SS in India, but one study has proved the rarity of SS in India.[4] In contrast, the prevalence of SS throughout the world is estimated to be 0.5% to 1%.[5] PM can occur years after the person has SS, which is a more common presentation, but it is sometimes present along with SS on its first presentation.[6]
Our patient suffered from the symptoms of both diseases simultaneously. EULAR gives guidelines for the treatment of both disorders. The recommendations for the management of SS are shown in Figure 1. There are guidelines by the British Society for Rheumatology about treating myositis.[7]
The presence of SSB/LA supports the diagnosis of SS, and several studies have shown the presence of SSB/LA and SSA together in patients with SS and PM and some cases of overlap syndrome. Our patient faced several issues due to a lack of proper diagnosis by the doctors and, later on, her noncompliance with medicines and follow-ups. However, one study suggests that the presence of antibody SSA/Ro52, which was present in our patient, and SSA/Ro60 are associated with resistance against the treatment. Due to the patient’s noncompliance, we cannot confirm the real reason behind the persisting symptoms. It was seen that the patient responded to the treatment initially, but more clarity could be gained on future follow-ups.
Our case report holds importance in terms of the epidemiology of SS and gives an idea to physicians to consider PM when assessing a patient of SS with musculoskeletal complaints.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of Patient Consent
The authors certify that they have obtained all appropriate patient consent forms. In the form the patient(s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
CRediT Author Statement
All authors contributed to concept & design, data collection & interpretation, drafting & revising, and approval for publication of this manuscript.
Data Availability
Nil.
Use of Artificial Intelligence
Artificial Intelligence was not used.