
Abstract

Dear Editor,
Immunoglobulin G4-related disease (IgG4-RD) is an immune-mediated fibro-inflammatory disorder characterised by tumefactive lesions, lymphoplasmacytic infiltration, and multiorgan involvement. 1 Overlap with idiopathic inflammatory myositis (IIM) is extremely uncommon and may lead to diagnostic confusion when associated with hypergammaglobulinemia mimicking monoclonal gammopathy. 2
A 60-year-old lady presented with 8 months of myalgia and 6 months of symmetrical proximal muscle weakness (Medical Research Council grade 4/5 upper limbs, 3/5 lower limbs) with weakness of neck and truncal muscles. She also had bilateral submandibular gland enlargement and a dry cough for 4 months without fever, joint pains, rash, raynaud’s or sicca symptoms. Laboratory investigations revealed raised inflammatory markers, like C-reactive protein (CRP) 99 mg/L, erythrocyte sedimentation rate (ESR) 58 mm in the first hour, and serum ferritin 2,200 µg/L. Muscle enzymes were raised with Creatine phosphokinase (CPK) 323 U/L (cut-off 170 U/L), aspartate transaminase (AST) 76 U/L, and alanine transaminase (ALT) 48 U/L. Lactate dehydrogenase (LDH) was 542 U/L, and aldolase 18 U/L, with low serum albumin 2.3 g/dL and elevated globulin levels 4.6 g/dL.
Differentials included disseminated tuberculosis, paraneoplastic myositis, Primary Sjögren’s Syndrome, IgG4-RD, and sarcoidosis. Sputum examination for Mycobacterium tuberculosis came negative. Neck ultrasonography showed enlarged submandibular glands, heterogeneous parotids, and cervical lymphadenopathy. Abdominal ultrasonography revealed bilateral grade 2 hydronephrosis. High-resolution computed tomography (HRCT) of the thorax showed pulmonary nodules with peribronchovascular and interstitial thickening, alongside ground-glass opacities (GGOs). Magnetic resonance imaging (MRI) of the thigh (STIR sequence) demonstrated diffuse muscle oedema suggestive of inflammatory myositis.
Myositis immunoblot revealed 3+ anti-PM-Scl positivity. Tumour markers were negative. Schirmer-I test and anti-Ro/La antibodies also turned out to be negative, making Primary Sjögren’s Syndrome unlikely. Serum angiotensin converting enzyme (ACE) was normal. Fine-needle aspiration cytology (FNAC) of the cervical lymph node showed reactive changes. Serum IgG4 level was elevated at 343 mg/dL (cut-off of 135 mg/dL). Serum protein electrophoresis (SPEP) revealed an M-spike in the gamma region. Serum kappa and lambda light chain levels were elevated at 76.01 mg/L (3.3-19.4 mg/L) and 78.87 mg/L (5.7–26.3 mg/L), respectively. She also had elevated serum β2-microglobulin levels at 7,057.68 ng/mL (800-2,340 ng/mL). These parameters raised suspicion of a monoclonal gammopathy.
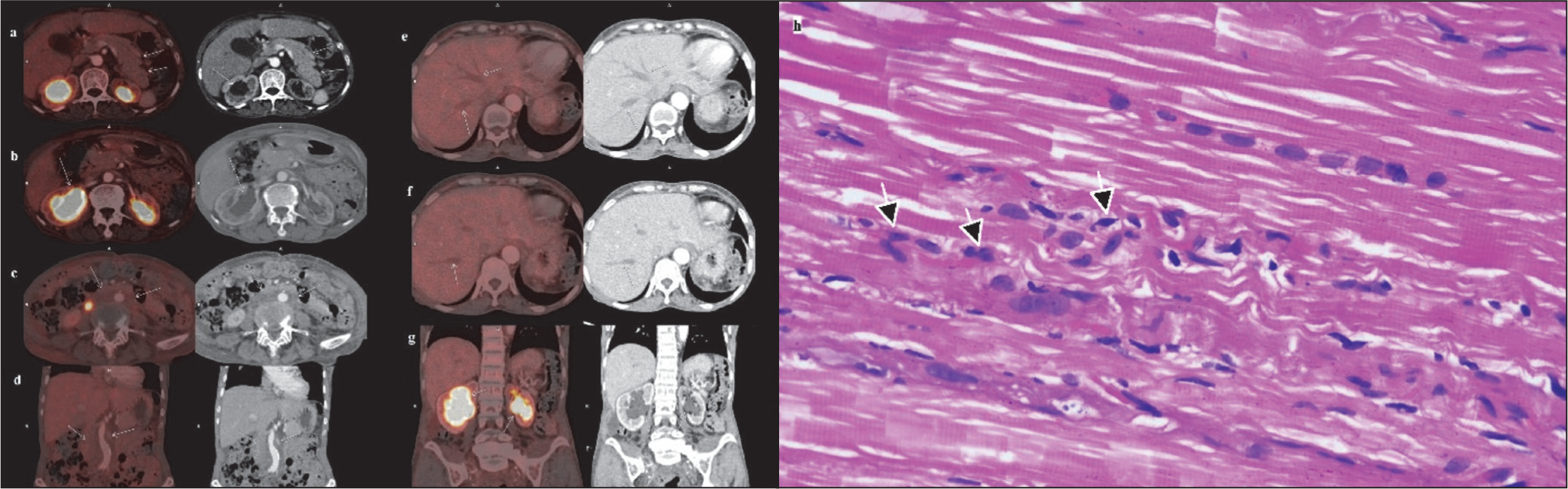
Fluorodeoxyglucose-Positron emission Tomography (FDG-PET) demonstrated a metabolically inactive bulky pancreas, retroperitoneal soft tissue thickening encasing the abdominal aorta, bilateral hydronephrosis and dilated intrahepatic biliary ducts (Figure 1a-1g) along with metabolically active pulmonary nodules (SUV-max 2.25), peribronchovascular, interstitial, and interseptal thickenings, with GGOs. Erdheim-Chester disease (ECD) was also considered a possibility along with IgG4-RD. The presence of retroperitoneal fibrosis with coated aorta, interstitial lung disease (ILD), and raised serum IgG4 levels was a pointer for ECD. However, absence of osteosclerotic lesions in the meta-diaphyses of long bones of lower limbs (usually present in > 95% cases of ECD), contrast-enhancing or hypermetabolic perinephric infiltrates, or involvement of endocrine organs made the possibility of ECD less likely. 3 Muscle biopsy revealed endomysial inflammation consistent with inflammatory myositis (Figure 1h) with negative IgG4 staining, confirming true disease overlap. Bone marrow showed reactive plasmacytosis (9%) without clonal restriction. Moreover, serum immunofixation electrophoresis done later refuted the presence of any monoclonal proteins.
Fluorodeoxyglucose Positron Emission Tomography Scan with Arrows Showing Bulky Pancreas with Loss of Lobulations (a), Dilated Renal Pelvis (Right More Than Left) with Increased Tracer Uptake (b, g), Metabolically Inactive Retroperitoneal Fibrous Tissue Encasing the Infrarenal Aorta (c, d), and Dilated Intrahepatic Bile Ducts and Radicles (e, f), and Muscle Biopsy From Right Vastus Lateralis Stained with Haematoxylin and Eosin (H&E) Showing Endomysial Inflammation with Infiltrates of Mononuclear Cells (Arrows) with Minimal Myocyte Necrosis and Regeneration (H&E, ×400) (h).
She fulfilled the 2019 American College of Rheumatology/European League Against Rheumatism (ACR/EULAR) inclusion criteria for IgG4-RD (Score 52; cut-off ≥ 20) and 2017 EULAR/ACR diagnostic criteria for IIM (Score 9.9; cut-off ≥ 8.7). The patient fitted into the classification criteria of IgG4-RD, after rationally ruling out all possible mimickers. However, we failed to provide any biopsy evidence from the organs consistent with IgG4-RD lesions. This was primarily because the patient refused further invasive evaluation after undergoing a muscle and bone marrow biopsy. We diagnosed her as a possible IgG4-RD with inflammatory myositis overlap. She was put on oral prednisolone 0.6 mg/kg/day and rituximab, leading to marked clinical improvement and a reduction of prednisolone dose to 5 mg/day within 4 months. She is presently doing well with the resolution of submandibular gland enlargement, marked improvement of respiratory symptoms, and proximal muscle weakness after 9 months of follow-up. Following the receipt of her first maintenance dose of rituximab at 6 months, her prednisolone dose was further tapered to 2.5 mg/d, with no new symptoms appearing since the taper. This also provided therapeutic evidence of an excellent response of an IgG4-RD to glucocorticoids and B-cell depleting therapy.
This overlap of IgG4-RD with inflammatory myositis is a rare one, with only one such case reported before. 4 Although IgG4-RD can rarely involve skeletal muscle, 5 absence of IgG4-positive plasma cells on biopsy confirmed coexisting IIM rather than IgG4-related myositis. Polyclonal IgG4 expansion may cause hypergammaglobulinemia and a band bridging β and γ fractions on SPEP, mimicking monoclonal gammopathy. 6
IgG4-RD can rarely coexist with IIM and masquerade as monoclonal gammopathy. A systematic clinico-radiological and histopathological approach is crucial for accurate diagnosis and management.
Footnotes
Authors’ Contribution
MM: conception and design of work, acquisition, analysis, and interpretation of data, drafting the work, final approval of the version to be published. RKH: acquisition of data, reviewing it critically for important intellectual content, final approval of the version to be published. KB: conception and design of work, analysis of data, reviewing it critically for important intellectual content, final approval of the version to be published. RE: acquisition, analysis, and interpretation of data, reviewing it critically for important intellectual content, final approval of the version to be published, DC: analysis, and interpretation of data, reviewing it critically for important intellectual content, final approval of the version to be published.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Ethical Approval
Since this is a case report, approval was not mandatory as per the IRB.
Funding
The authors received no financial support for the research, authorship and/or publication of this article.
Patient Consent
Informed consent was obtained from the patient for publication.