
Abstract

Dear Editor,
An adolescent male presented with a five-month history of insidious-onset, erythematous, non-pruritic malar rash that progressively evolved into painful, indurated nodular lesions over both cheeks. The rash was accompanied by diffuse facial swelling and periorbital oedema, resulting in restricted eyelid opening (Figure 1A). He was initially treated empirically with antihistamines and oral corticosteroids (prednisolone 0.5 mg/kg/day) for suspected angioedema, with partial improvement; however, symptoms recurred following cessation. Two weeks before presentation, he developed high-grade, continuous fever (102-103 °F) with chills and rigours, unresponsive to antipyretics and without localising features. There was no personal or family history of atopy, autoimmune disease or drug use. Examination revealed firm, erythematous, indurated malar swelling, periorbital oedema, discrete non-tender cervical lymphadenopathy and pallor.
(A) Hard Indurated Swelling of Bilateral Cheek with Periorbital Oedema at Presentation; (B) Sustained Clinical Remission at 12-month Follow-up Visit; (C) Haematoxylin and Eosin, ×200: Section Shows Adipose Tissue with Mildly Enlarged Lymphoid Cells Rimming the Adipocytes (Arrow); (D) Diaminobenzidine, ×200: Numerous CD3 Positive T Lymphoid Cells Rimming the Adipocytes (Arrow); (E) Diaminobenzidine, ×200: Adipose Tissue with Only Few Scattered CD20 Positive B Lymphoid (Arrow).
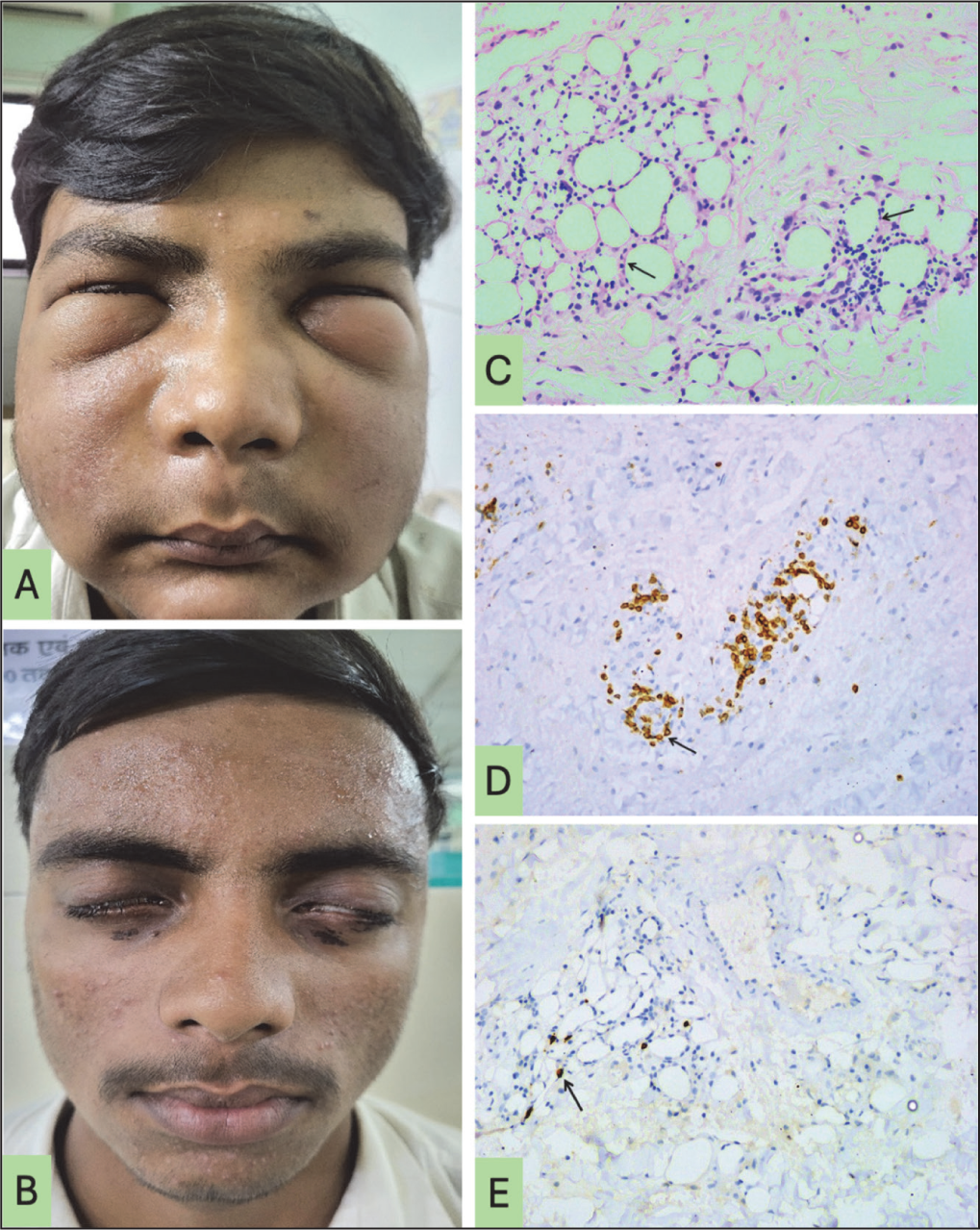
Initial evaluation revealed pancytopenia (haemoglobin 8.6 g/dL, total leukocyte count 2,340/mm³, platelets 1.1 × 10⁵/mm³), with disproportionately elevated C-reactive protein (12 mg/dL) and low erythrocyte sedimentation rate (2 mm/h). Liver function tests showed mild transaminitis (serum glutamic oxaloacetate transaminase [SGOT] 65 IU/mL, serum glutamic pyruvate transaminase [SGPT] 32 IU/mL), while renal function and thyroid profile were within normal limits. Notable abnormalities included elevated serum lactate dehydrogenase (LDH) (1,677 IU/mL), hyperferritinaemia (594 ng/mL), hypertriglyceridaemia (236 mg/dL) and markedly elevated serum immunoglobulin E (IgE) (1,535 IU/mL). Infectious workup and autoimmune serologies (anti-nuclear antibody [ANA], extractable nuclear antigen (ENA) panel, anti-ds deoxyribonucleic acid [DNA]) were negative. Ultrasonography revealed splenomegaly and bilateral cervical and intra-parotid lymphadenopathy. Magnetic resonance imaging (MRI) of the face and neck demonstrated diffuse T2/short tau inversion recovery (STIR) hyperintensities involving the subcutaneous and muscular planes of the buccal and facial regions, suggestive of an infiltrative process with associated panniculitis and focal myositis. Bone marrow examination showed normoblastic erythroid hyperplasia with a normal myeloid-to-erythroid ratio (1:1) and no evidence of haemophagocytosis. Histopathological examination of the subcutis and left masseter muscle biopsy revealed moderate lobular panniculitis with lymphohistiocytic infiltration, foci of necrosis and adipocyte rimming (Figure 1C). Immunohistochemistry (IHC) demonstrated cluster differentiation (CD)3-positive atypical T-cell infiltrates with scattered CD20-positive B cells, consistent with a diagnosis of SPTCL (Figure 1D and E). Further, immunohistochemical subtyping for T-cell clonality could not be performed as these facilities were unavailable at our institution and referral for external IHC evaluation was deferred owing to financial constraints.
In view of impending macrophage activation syndrome secondary to SPTCL, the patient was initiated on intravenous pulse methylprednisolone (500 mg/day for three consecutive days), followed by a tapering regimen of oral prednisolone at 0.5 mg/kg/day along with a combination of cyclosporine. Progressive reduction of facial and periorbital oedema along with laboratory improvement was documented as early as three weeks and maintained a sustained remission up to 12-month follow-up (Figure 1B). Of note, written consent was taken from the patient for the publication.
Panniculitis, an inflammatory disorder of subcutaneous adipose tissue, encompasses a broad differential including infectious, autoimmune and autoinflammatory aetiologies. However, facial panniculitis is distinctly uncommon and typically restricted to specific conditions such as cold-induced panniculitis, lupus panniculitis, dermatomyositis, localised infections, trauma, sarcoidosis and SPTCL, each necessitating a meticulous and thorough diagnostic approach.
Subcutaneous panniculitis-like cytotoxic T-cell lymphoma is a distinct clinical entity which shows clonal lymphocytic proliferation. Clinically, it presents with erythematous painless nodules or plaques in limb, trunk and rarely in the face, with or without accompanying systemic symptoms. It follows a relapsing and remitting course with lesions at different stages of healing. Although markedly elevated serum IgE is not a defining feature of SPTCL (unlike Sezary syndrome), it may be observed across T-cell lymphoproliferative disorders, reflecting IL-4-driven IgE induction by activated T-cell clones rather than disease specificity. 1 Typical histology reveals lobular panniculitis with classical peri-adipocyte rimming of clonally proliferated T-cells. Infiltrating cells are mostly cytotoxic CD8+ cells expressing the cytotoxic proteins TIA1, granzyme B and perforin, but not CD56. Immunohistochemical analysis confirms the lymphocyte clone with specific T cell receptor (TCR) gene rearrangements (ab or gd). 2 Prognosis is usually excellent in young adults but may vary in patients with Hepatitis A virus cellular receptor (HAVCR)2 mutation (encoding T cell immunoglobulin and mucin domain [TIM-3]) with hemophagocytic lymphohistiocytosis (HLH)-like presentation. Diagnosing orofacial lymphomas remains challenging due to their clinical overlap with conditions such as lupus panniculitis and cellulitis. Management typically involves high-dose corticosteroids, immunotherapy and cyclosporine.
Footnotes
Authors’ Contribution
The conceptualisation, study design, acquisition of data, analysis by RKS, SM; interpretation of data, manuscript writing, editing and final draft approval by RKS, SM, PK, KPM. Supervision and revision of the manuscript by PK, KPM. All authors to be accountable for all aspects of the work in ensuring its accuracy or integrity.
Data Availability
Data will be made available upon reasonable request to the first or corresponding author of the study.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Ethical Approval Statement
Not required as case report and consent was taken directly from the patient.
Funding
The authors received no financial support for the research, authorship and/or publication of this article.
Patient Consent
Written informed consent was obtained from the patient before the publication and is attached.