
Abstract
Objective:
To describe the clinical profile and outcome of patients with catastrophic antiphospholipid syndrome (CAPS) from a tertiary care Rheumatology centre in North India.
Methods:
A retrospective analysis of data of patients between 1990 and March 2024 was done. Those with rapid involvement of three or more organ systems with moderate to high titres of antiphospholipid antibodies (aPL) at least once were considered physician-diagnosed CAPS.
Results:
Fourteen patients were considered as cases of CAPS. The majority were females, with a median age of 28 years (26.25–37.25). Two-thirds of the patients had systemic lupus erythematosus (SLE) as the background illness. Thrombocytopenia was present in all cases, and CAPS was the presenting manifestation in half of them. An identifiable trigger was found in only nine cases predominantly infection. Triple therapy with intravenous immunoglobulin (IVIG), pulse steroids, and heparin was received by six patients, resulting in clinical recovery in five of them. Nine patients received anticoagulation and the rest received glucocorticoids, other immunosuppressive agents and antibiotics for underlying sepsis. Among the five patients who did not receive heparin, three died of multiorgan dysfunction. Two patients died despite anticoagulation owing to late presentation and delay in initiation of heparin.
Conclusion:
A high index of suspicion should be kept for CAPS, especially in a young female with rapidly progressive multiorgan dysfunction in the background of autoimmune features or pregnancy morbidity. Triple therapy using heparin, IVIG and high-dose steroids should be considered in patients with suspected CAPS.
Introduction
Antiphospholipid syndrome (APS) is characterised by vascular thrombosis and pregnancy morbidity in the presence of antiphospholipid antibodies (aPL). Catastrophic APS (CAPS) represents a rare complication of APS, characterised by the involvement of multiple vascular beds defined by rapid involvement of three or more organ systems within a span of one week, occurring in less than 1% of patients. 1 It is a close differential diagnosis of disseminated intravascular coagulation (DIC) and thrombotic thrombocytopenic purpura (TTP). In addition, coexisting DIC is observed in up to 15%-20% of CAPS cases2,3 also referred to as Asherson syndrome. 4
CAPS exhibits a strong female preponderance, with nearly half of the patients having primary APS and another 40% having CAPS secondary to systemic lupus erythematosus (SLE). 2 It may be the presenting manifestation of APS in up to half the patients and 40%-50% have identifiable triggers such as sepsis, lupus disease activity, pregnancy, trauma, malignancy or withdrawal of anticoagulation.5,6
The kidney, lung, brain and heart are the most common organs involved while thrombocytopenia is very common. 2 CAPS should be suspected when two or more organs are involved in rapid succession in the setting of either APS or appropriate triggers. Typically, patients have elevated serum creatinine, lung involvement with ARDS and/or diffuse alveolar haemorrhage (DAH), encephalopathy from microvascular disease or stroke, heart failure from myocardial dysfunction or infarct, hepatic dysfunction or cutaneous infarcts. Cardiopulmonary involvement indicates a poor prognosis. 7
Organ dysfunction in CAPS results from widespread microvascular thrombosis, although large vessel occlusions can also occur, 8 and thrombocytopenia may be severe with prolonged activated partial thromboplastin time (aPTT) and normal fibrinogen. 9 There is a systemic inflammatory response and elevated cytokine levels like those seen in septic shock. 10 Proposed mechanisms underlying this immune thrombotic storm include alterations in the coagulation cascade, abnormal fibrinolysis,11,12 the role of toll-like receptors (TLRs), 13 widespread complement activation 14 and neutrophil extracellular traps (NETosis). 15
Early recognition and initiation of anticoagulation with therapeutic doses of heparin is lifesaving. In addition, identification and treatment of triggers such as infection, lupus disease activity, and supportive care are necessary. In CAPS associated with SLE triple therapy involving heparin, high-dose intravenous glucocorticoids, and intravenous immunoglobulins (IVIG) is beneficial.5,16,17 Additional therapies such as plasma exchange (PLEX), 18 B cell-depleting agents like rituximab 19 and complement inhibitors like eculizumab 20 have been tried. Despite aggressive treatment, mortality is still nearly 50%. 2
The clinical profile and outcome of CAPS patients have been reported in various cohorts and the largest data is available from the CAPS registry. 2 However data regarding the same is scarce, especially from the South Asian population. In this study, we aim to describe the clinical profile and outcome of patients with physician-diagnosed CAPS from a tertiary care Rheumatology centre in North India.
Methods
A retrospective analysis of electronic medical records and clinic files spanning from 1990 to March 2024 was conducted at a single centre, as per the guidelines of an institutional ethics committee. Patients who had rapid involvement of at least three organ systems within a period of less than 1 week and positive aPL antibodies in moderate to high titre or a positive lupus anticoagulant test (LAC) at least once, were considered as physician-diagnosed CAPS cases (Figure 1). The modified Sapporo criteria for APS, 21 2023 ACR/EULAR classification criteria for APS 22 and the preliminary classification criteria for CAPS 2 were further applied to these cases. Patients with missing data pertaining to APS antibody profile, other relevant investigations and treatment were excluded from the study.
Method for Selection of Cases.
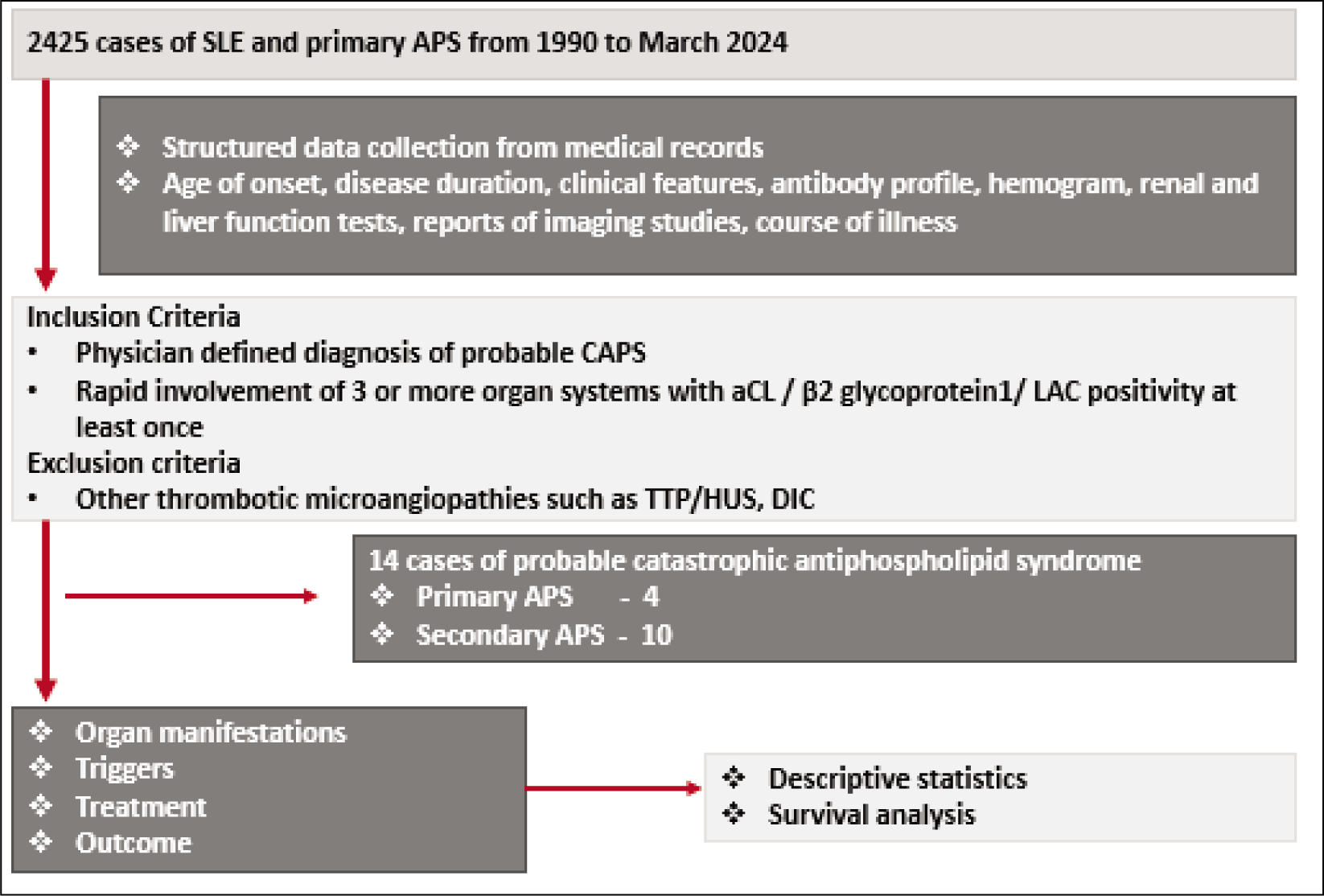
The demographic details as well as both criteria and non-criteria clinical features of APS were extracted for all patients. Antinuclear antibody tests (indirect immunofluorescence) and complement levels (nephelometry) were recorded when available. Anticardiolipin antibodies (aCL, IgM and IgG) and anti β2 glycoprotein 1 antibodies (total) were assessed via ELISA at our centre. LAC assays were conducted only when patients were not on anticoagulants. All cases displaying clinical features of SLE were categorised as secondary APS. An attempt to identify triggers was made for all probable APS cases.
Categorical data have been represented as percentages, while continuous data is presented as median with interquartile range (IQR). Survival analysis was done using R studio (Version 4.0.3) and a log-rank test was used for comparison.
Results
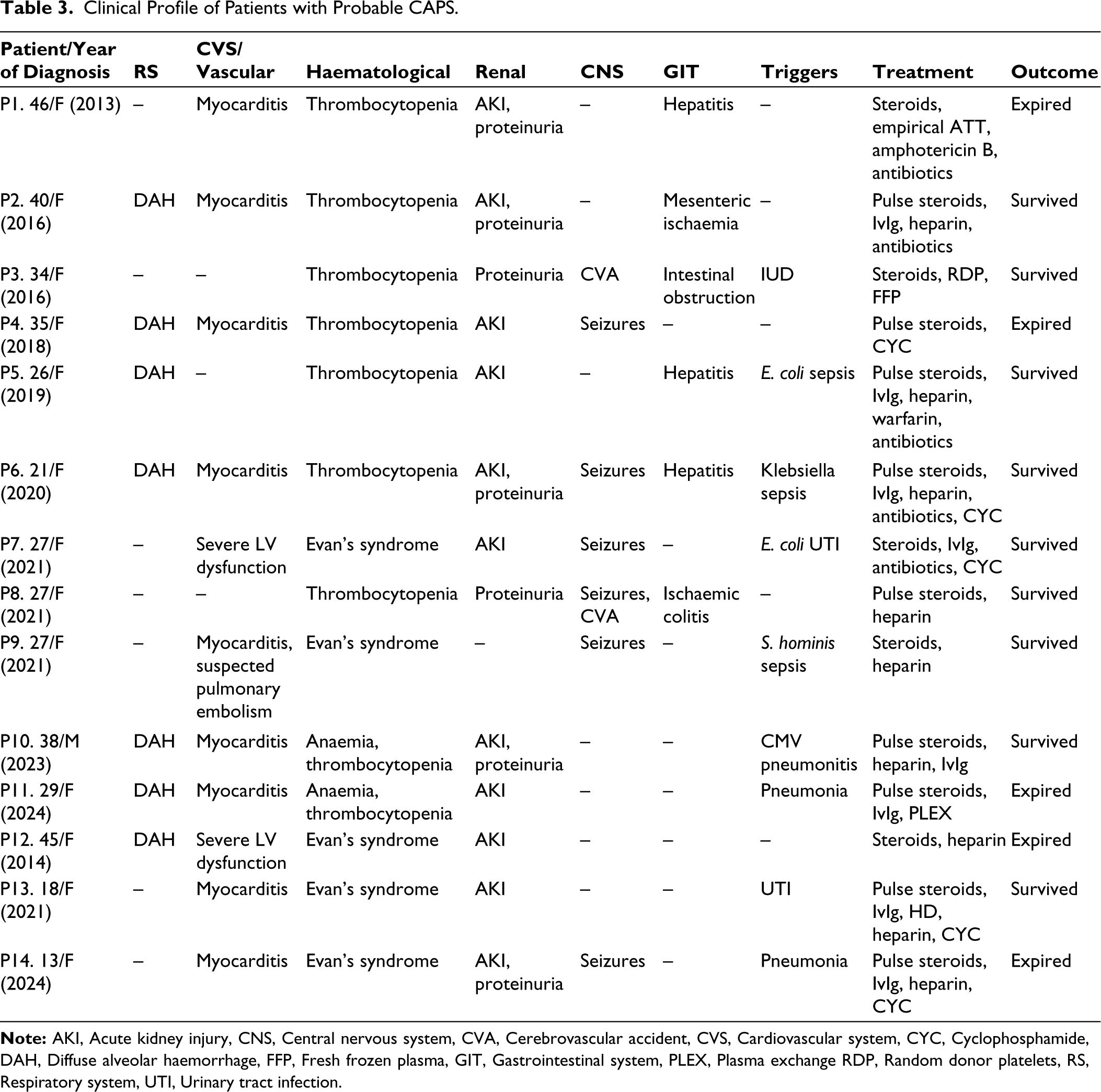
Fourteen cases of physician-diagnosed CAPS were identified. Out of these 14 cases, eight patients fulfilled both Sapporo and the ACR/EULAR criteria for APS. Confirmation of persistent aPL antibodies or LAC was not feasible in certain patients in view of anticoagulation or death. Preliminary criteria for probable CAPS were satisfied by only four patients as none of our patients underwent biopsy for histopathological evidence of thrombosis and imaging for thrombosis was done in four patients.
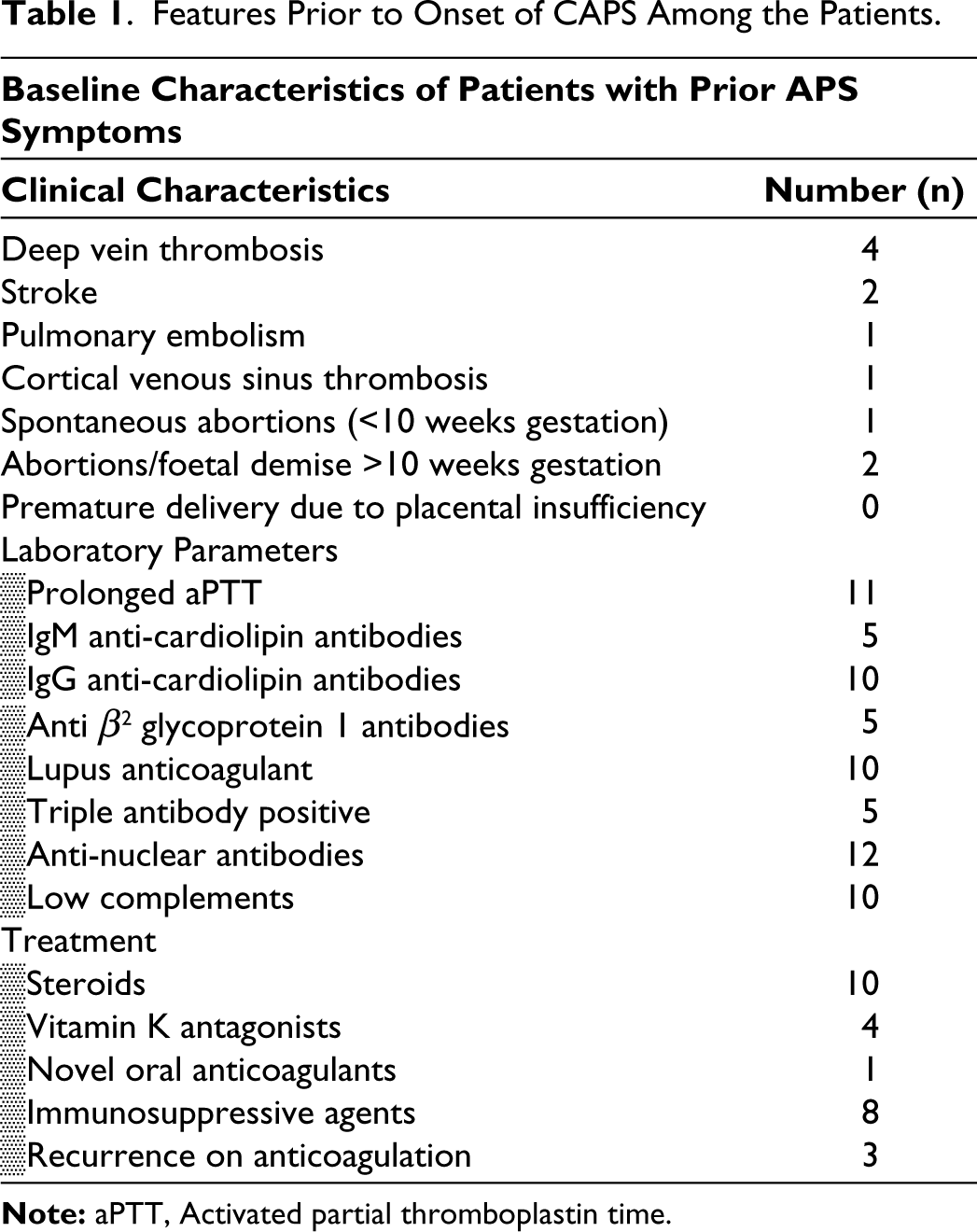
The median duration from first symptom to CAPS development was 1.5 years (0.2-5). In half of the patients, it was the initial manifestation of APS. Most were females (13/14), with a median age of 28 years (26-37) and 10 had SLE (Table 1).
Features Prior to Onset of CAPS Among the Patients.
Organ Involvement
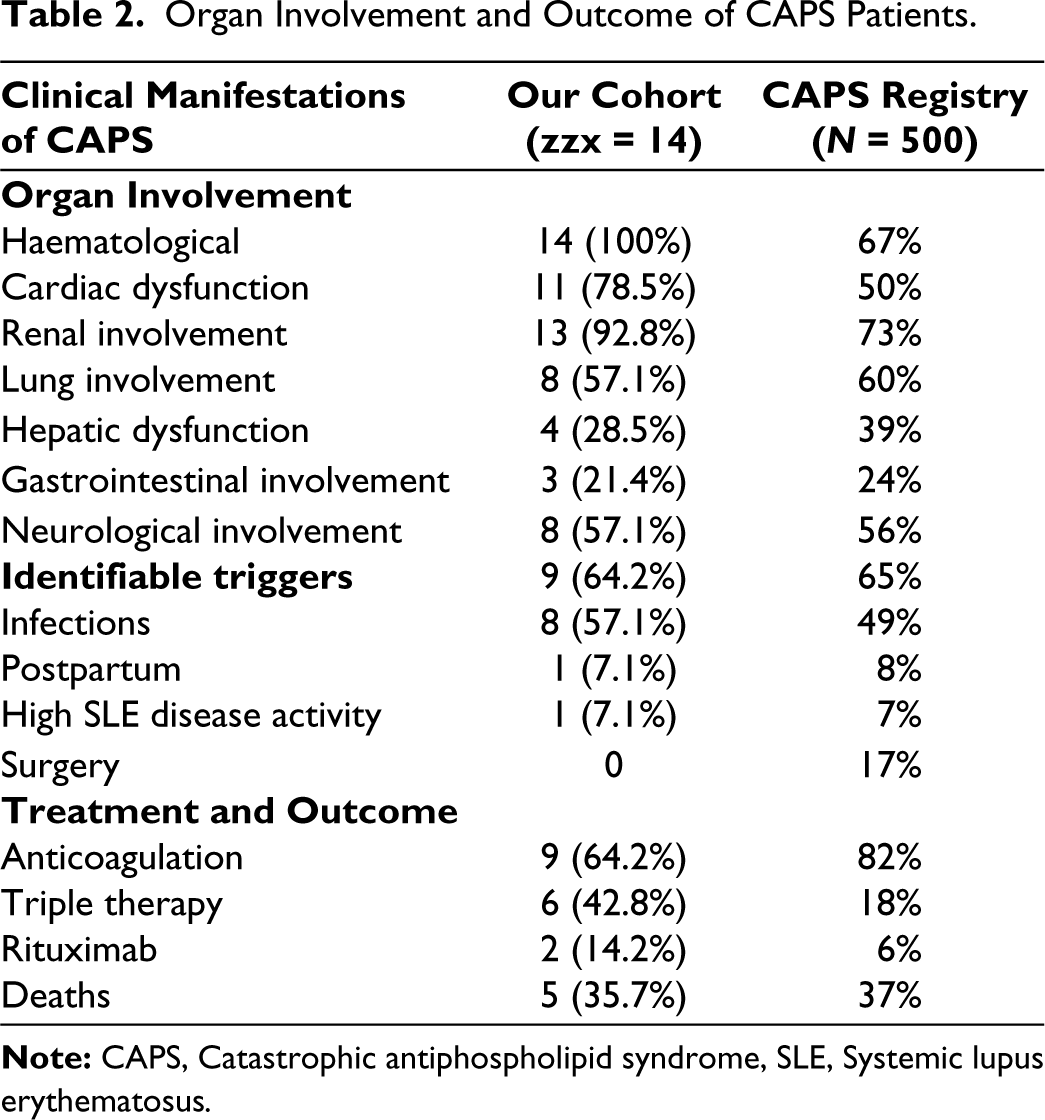
The common organ involvement was renal, cardiac and lung (Table 2). Rapidly progressive renal failure (RPRF) was seen in 11 patients. A renal biopsy was done in two patients, showing class IV+V lupus nephritis in one patient and minimal change of disease in the other. Myocarditis and echocardiographic findings indicating global left ventricular hypokinesia were seen in 10 patients. Pulmonary involvement occurred in eight patients, with DAH being the most common manifestation. Neurological and gastrointestinal involvement occurred in less than half of patients, with seizures noted in six and focal neurological deficits observed in two patients. Five patients had altered liver function, with hyperbilirubinemia in three and elevated transaminases in four patients. None exhibited AST/ALT levels > 1000 IU/mL. Concurrent deep venous thrombosis, Budd-Chiari syndrome, or digital gangrene were absent in all patients. Cytopenia was common with a median platelet count of 39,000/mm³ (21,500-57,500). Anaemia was present in all cases, while leukopenia occurred in four patients. The clinical features of each case are described in Table 3.
Organ Involvement and Outcome of CAPS Patients.
Clinical Profile of Patients with Probable CAPS.
Triggers
A potential trigger was identified in nine cases, with infection being the most common. Concurrent infections included Escherichia coli, Klebsiella and Staphylococcus hominis in three patients, while one patient had CMV pneumonitis and another had E. coli urinary tract infection. CAPS was triggered by intrauterine foetal demise (six months gestation) in a young female, which manifested 2 days post-partum. She had gestational hypertension but no features suggestive of pre-eclampsia. Another patient presented to our centre with DAH and renal impairment while he was on dabigatran following previous episodes of deep vein thrombosis and cerebral venous thrombosis prescribed from elsewhere. Elevated anti-dsDNA and hypocomplementemia were seen in one patient suggesting high disease activity which could have been a potential trigger for CAPS.
Treatment and Outcome
Triple therapy comprising IVIG, pulse steroid (0.25-1 gram per day for 1-3 days), and heparin was given to six patients, out of whom five patients survived. The total IVIG dose was 2 gm/kg, with one patient receiving low molecular weight heparin and the rest receiving therapeutic doses of unfractionated heparin. Nine patients received anticoagulation, and the rest received glucocorticoids, other immunosuppressive agents and antibiotics for underlying sepsis. One patient who was admitted to the nephrology unit with renal failure additionally received four cycles of PLEX. Two patients received rituximab following triple therapy as part of SLE management.
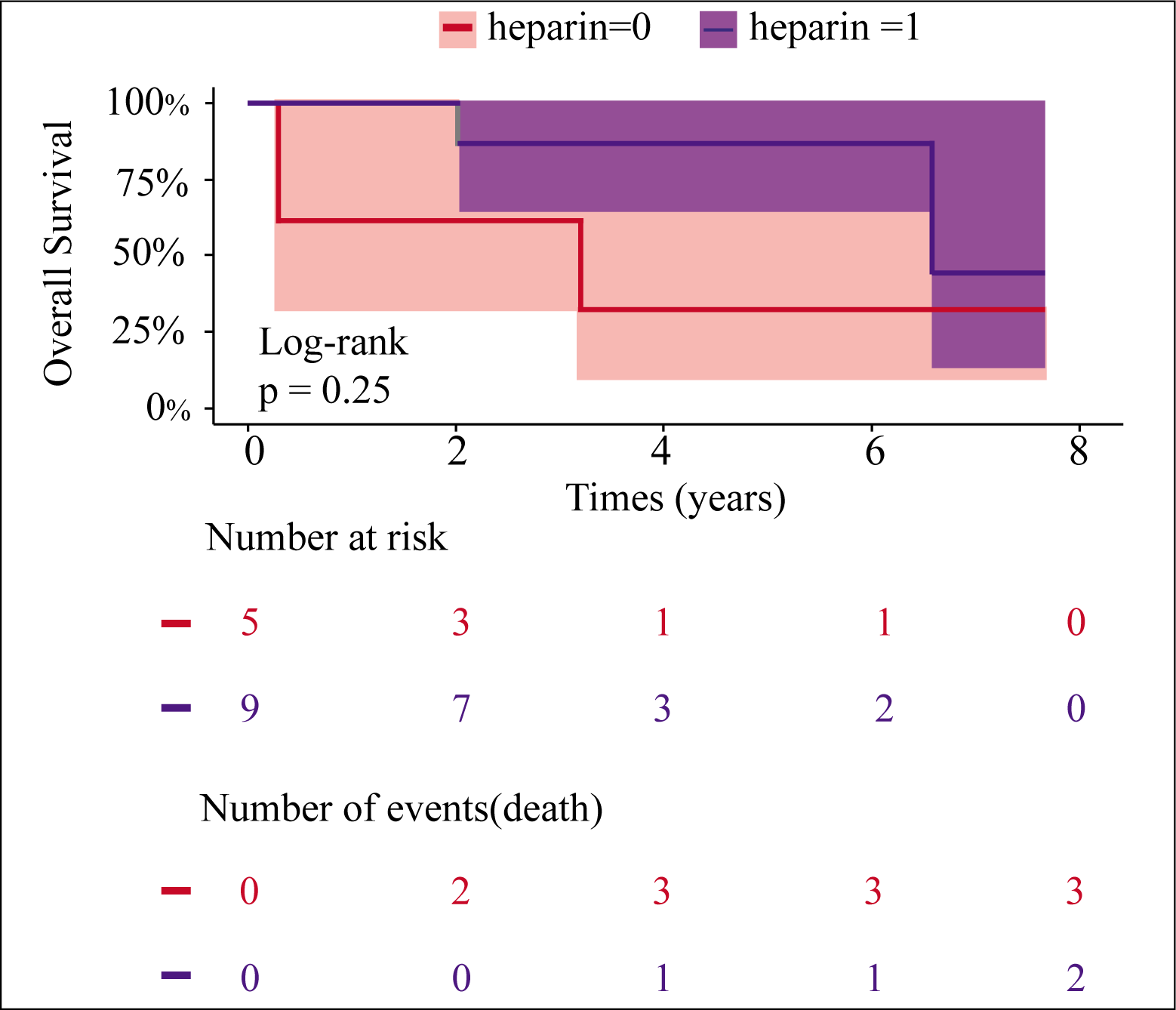
Among the five patients who did not receive heparin, three succumbed to multiorgan dysfunction (Figure 2). All survivors of CAPS were continued on anticoagulation with vitamin K antagonists. However, one patient died due to SLE disease activity during follow-up.
Kaplan-Meier Curve Showing Overall Survival.
Discussion
APS is a relatively rare autoimmune disease, with an estimated prevalence of 40-50 cases per 100,000 individuals, and catastrophic APS is even rarer. 1 The distribution of CAPS secondary to SLE in our cohort aligned with previous data (Table 3). Despite a slightly younger median age of 28 years, we observed a similar female predominance, consistent with findings from other studies.1,5,23 The most common aPL antibodies among our patients were LAC and IgG anticardiolipin antibodies, consistent with previous reports showing a high prevalence of IgG aCL antibodies in patients of Indian descent.24,25
A potential triggering factor was identified in two-thirds of the patients, with half of them developing CAPS following an infection, echoing findings from the CAPS registry. 26 Proposed role of infections in triggering CAPS include mechanisms such as molecular mimicry. 27 Structural similarities between microbes and β2 glycoprotein 1 can contribute to autoantibody formation. 28 Also following infections or other triggers, the conformational changes that occur can lead to exposure of the domain 1 of β2 glycoprotein1, 29 leading to an immune response. Additionally, inadequate anticoagulation can serve as another trigger, as seen in one of our patients who was triple antibody positive and developed CAPS while on dabigatran. Previous studies have shown an increased incidence of arterial thrombotic events and stroke with the use of novel oral anticoagulants (NOACs), especially in those with a high-risk aPL antibody profile. 30
Thrombocytopenia was present in all our patients, and organ involvement mirrored data from other cohorts in most domains (Table 3). However, we had more patients with seizures and the frequency of stroke was comparatively low in our patients. The multiorgan dysfunction associated with CAPS is associated with a diverse set of differential diagnoses including other thrombotic microangiopathies (TMA) like TTP, DIC, heparin-induced thrombocytopenia (HIT), sepsis with multiorgan dysfunction and haemolysis, elevated liver enzymes, low platelet count (HELLP) syndrome. 10 The very rapid onset of symptoms and clinical deterioration in the absence of anticoagulation are the key features that distinguish CAPS from other TMAs. Additionally, an elevated aPTT with normal prothrombin time favoured the diagnosis of probable CAPS over DIC in our patients.
Out of the 14 patients, nine had received anticoagulation, and six received triple therapy comprising high-dose glucocorticoids, IVIG/PLEX, and anticoagulation. Notably, all of these patients presented after 2015, suggesting improved management due to increased awareness and early recognition. B-cell-depleting therapies and drugs targeting the complement cascade have shown promise in the management of CAPS.19,20 Two of our patients received rituximab following triple therapy in view of the severe organ involvement (renal and severe thrombocytopenia) and are currently doing well.
Our study highlights good outcomes with the use of triple therapy and need for recognition of CAPS though it is often difficult to prove CAPS due to patients having multiorgan dysfunction with thrombocytopenia that precludes any biopsy and imaging studies needing contrast. Our data has multiple limitations including small number of patients, a lack of fulfilling classification criteria by all patients and a retrospective nature of data.
In conclusion, our data suggests that CAPS should be considered in young females with multiorgan dysfunction and early use of heparin can be lifesaving. In future, a prospective multicentric study from India is needed to know the triggers and outcomes of CAPS in our setting.
Footnotes
Authorship Contributions
Conceptualisation: LMR, KNT, AA.
Methodology: All authors.
Formal analysis and investigation: LMR, KNT.
Writing - Original draft preparation: LMR, KNT.
Writing - Review and editing: All authors.
Funding acquisition: No funding received.
Supervision: DPM, VA, AL, AA.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Ethics Approval
No IEC approval was taken as it is a retrospective case series.
Funding
The authors received no financial support for the research, authorship and/or publication of this article.
Informed Consent
This was a retrospective review of case records; hence written consent was not obtained from all patients. Confidentiality of patient data was maintained.