
Abstract
Objective:
The objective of this study is to evaluate the efficacy and safety of tofacitinib in patients with ankylosing spondylitis (AS).
Methods:
In this randomised, double-blind, placebo-controlled study, patients with active AS aged between 18 and 60 years who fulfilled the modified New York criteria for AS and had inadequate response or intolerance to non-steroidal anti-inflammatory drugs were enrolled. Patients were randomised to receive either tofacitinib 5 mg twice daily or placebo for 12 weeks. The primary endpoint was Assessment of Spondyloarthritis International Society ≥20% improvement (ASAS20), and the secondary endpoints included Assessment of Spondyloarthritis International Society ≥40% improvement response, mean change from baseline in Bath Ankylosing Spondylitis Disease Activity Index (BASDAI), Ankylosing Spondylitis Disease Activity Score–C-reactive protein (ASDAS–CRP), Bath Ankylosing Spondylitis Metrology Index (BASMI), Bath Ankylosing Spondylitis Functional Index (BASFI), Maastricht Ankylosing Spondylitis Enthesitis Score (MASES), erythrocyte sedimentation rate (ESR), C-reactive protein (CRP), serum tumour necrosis factor alpha (TNF-α) and interleukin 17A (IL-17A) levels. Safety assessment was done throughout the study period.
Results:
Forty-eight patients were included in the study and received either tofacitinib (n = 25) or placebo (n = 23). The ASAS20 response was significantly higher in the tofacitinib group compared with placebo (68% vs 13%, P < .001). Significant improvement was also seen in secondary endpoints, including mean change in BASDAI (P < .001), ASDAS–CRP (P < .001), BASMI (P = .003), BASFI (P = .001), ESR (P = .002), CRP (P = .003), serum TNF-α (P = .007) and IL-17A levels (P <.001). There was no significant difference in MASES (P = .7). Both groups had similar infection rates.
Conclusions:
In patients with active AS, tofacitinib was effective in reducing disease activity measures compared with placebo.
Introduction
Ankylosing spondylitis (AS), characterised by inflammation affecting the axial skeleton, has a chronic, occasionally relentless inflammatory course resulting in significant deterioration in quality of life.1–3 Non-steroidal anti-inflammatory drugs (NSAIDs) are considered the first line of treatment for AS. Non-pharmacological measures, such as patient education and physiotherapy, are also often advised.4,5 Biologic disease-modifying antirheumatic drugs (bDMARDs) are indicated for patients with active disease despite NSAID therapy or for whom NSAIDs are contraindicated or not tolerated as conventional disease-modifying antirheumatic drugs are often ineffective.4–7 Intolerance or inadequate response to bDMARDs is reported in around 20%–40% of patients with AS. 8 Consequently, there is an unmet need for alternative oral treatments that are convenient to use and have possibly different mechanisms of action.
Tofacitinib, an oral Janus kinase (JAK) inhibitor, holds significant potential in the treatment of AS. It inhibits JAK1 and JAK3 enzymes and blocks downstream signalling of several pro-inflammatory cytokines.9,10 The interleukin (IL)-23/IL-17 pathway plays a major role in the pathogenesis of axial spondyloarthritis (axSpA) with higher serum levels of IL-23 and IL-17 reported in AS. 11 IL-17, by activating osteoclasts, inhibits bone formation. It orchestrates with tumour necrosis factor-alpha (TNF-α) and interferon-γ and contributes to the structural damage in AS. Hence, JAK inhibitors could potentially reduce inflammation and alleviate symptoms in AS.
Phase 2 and 3 trials of tofacitinib in AS showed greater clinical efficacy vs placebo with regard to symptom improvement and reducing disease activity.12,13 The magnetic resonance imaging outcomes at 12 weeks, including Berlin score, Spondyloarthritis Research Consortium of Canada (SPARCC) scores of the sacroiliac joints and the spine, were also improved. 13 Upadacitinib, an oral selective JAK1 inhibitor, was assessed in biologically naive patients with AS who had persistent symptoms despite NSAID treatment (SELECT-AXIS 1) and demonstrated significant improvement in objective endpoints compared to placebo (P = .0003). 14 Upadacitinib was also effective in non-radiographic axial SpA vs placebo (SELECT-AXIS 2). 15 Filgotinib, another selective JAK1 inhibitor, showed significant clinical efficacy in patients with active AS (TORTUGA). 16
The present study evaluated the efficacy and safety of tofacitinib in patients with active AS and its correlation with serum IL-17A and TNF-α levels.
Materials and Methods
This single-centre, randomised, double-blind, placebo-controlled study was conducted in the Department of Clinical Immunology and Rheumatology, Institute of Medical Sciences and Siksha ‘O’ Anusandhan (SUM) Hospital, Bhubaneswar, India, from June 2022 to May 2023.
Randomisation and Sequence Allocation
Randomisation of patients was done by a computer-generated (
Ethical Clearance
Institutional ethical clearance was obtained before starting the study, and the trial was registered under the Clinical Trials Registry, India.
Study Groups
Patients were allocated either tofacitinib 5 mg twice daily or placebo. Drug compliance was considered adequate if 80% of the drug was consumed. Drugs such as NSAIDs, sulfasalazine at a dose of ≤3 g/day, methotrexate at a dose
of ≤25 mg/week and oral steroids not more than 10 mg/day of prednisone or another steroid in equivalent doses were allowed to continue throughout the study without any alteration in dosage. A stable dose had to be maintained for at least 4 weeks for sulfasalazine or methotrexate and 1 week for oral steroids before enrolment. Injected (intraarticular/intramuscular) corticosteroids were to be discontinued 4 weeks before inclusion in the study. Non-pharmacological therapies were continued as indicated.
Patients
Patients selected for the study were adjudged to have fulfilled the following criteria: (a) age between 18 and 60 years fulfilling the modified New York criteria for AS (1984), 17 (b) BASDAI ≥4, (c) intolerance or inadequate response to at least two different oral NSAIDS for a total duration of at least 4 weeks and (d) patient not affording bDMARDs, including TNF and IL-17 inhibitors. Written informed consent was taken from all participants.
The exclusion criteria included (a) patients previously exposed to or current treatment with JAK inhibitors or bDMARDS; (b) blood dyscrasias at screening—haemoglobin <8 g/dl, absolute white blood cell count <3,000 per mm3 and platelet count <100,000 per mm3; (c) renal insufficiency—creatinine clearance <40 ml/min, calculated using Cockcroft–Gault formula; (d) liver insufficiency—bilirubin and liver enzymes ≥1.5 upper limit of normal at screening; (e) complete ankylosis of the spine; (f) patients having active tuberculosis or positive for latent tuberculosis as tested by QuantiFERON-TB Gold In-Tube (QFT Gold test) or Mantoux test; (g) history of recurrent herpes zoster or herpes simplex; (h) present malignancy or with a history of malignancy; (i) current or past history of thromboembolism or presence of risk factors for the same; (j) history of coronary artery disease or presence of risk factors for cardiovascular disease like diabetes mellitus, hypertension and dyslipidaemia; and (k) pregnant female or breast-feeding mothers.
A phase 3 trial of tofacitinib in AS (randomised and placebo-controlled) showed that the Assessment of Spondyloarthritis International Society 20 (ASAS20) response was achieved in 56.4% of patients receiving tofacitinib and 29.4% in those who received placebo at 16 weeks. 13 To detect this difference at a significant level of 5% (P < .05) and 80% power, the calculated sample size was 98. Hence, 100 patients were planned to be randomised into two equally divided groups to receive oral tofacitinib and placebo. However, 48 patients could be recruited during this time period and were randomised in the study (tofacitinib group = 25 patients and placebo group = 23 patients).
Main Outcome Variable
At week 12, an ASAS20 response of ≥20% and at least a unit’s improvement from baseline in three out of four components and no worsening of ≥20% and ≥1 unit in the remaining component was assessed. ASAS20 response scores patient global (numerical rating scale 0–10), pain (numerical rating scale 0–10), function (Bath Ankylosing Spondylitis Functional Index [BASFI]) and inflammation, which is the mean of Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) questions 5 and 6.
Other Variables
At week 12, an Assessment of Spondyloarthritis International Society 40 (ASAS40) response of ≥40% and at least two units improvement in ≥3 of 4 components compared to baseline, as well as no worsening of ≥40% and ≥2 units in the other component, mean change from baseline to week 12 in BASDAI, Ankylosing Spondylitis Disease Activity Score–CRP (ASDAS–CRP), Bath Ankylosing Spondylitis Metrology Index (BASMI), BASFI, Maastricht Ankylosing Spondylitis Enthesitis Score (MASES), Swollen Joint Count 44 (SJC44), erythrocyte sedimentation rate (ESR), c-reactive protein (CRP), serum TNF-α and IL-17A levels were measured. A continuous assessment of safety was also performed throughout the study period.
Procedures
A detailed history was elicited from all participants who also underwent a thorough physical examination. Data were collected regarding age, sex, duration of onset of symptoms and diagnosis, current or past history of peripheral articular involvement, psoriasis, inflammatory bowel disease and uveitis, thromboembolic episodes, contact with tuberculosis and details of treatment received. Laboratory investigations included complete blood count, renal and liver function tests, HBsAg, anti-HCV and HIV antibodies, ESR and CRP. Radiographs of the pelvis anteroposterior view, lumbar spine anteroposterior and lateral views and chest posteroanterior view were done. Disease activity assessment was done using BASDAI, BASFI, BASMI and SJC44. Serum IL-17 and TNF-α levels were measured by the enzyme-linked immunosorbent assay method according to manufacturer instructions. ASAS20 and ASAS40 responses were assessed at 12 weeks. BASDAI, ASDAS–CRP, BASMI, BASFI, SJC 44, MASES, ESR and CRP were assessed at participant induction and every 4 weeks thereafter. Serum IL-17A and TNF-α levels were assessed initially and then at week 12. The study protocol is shown in Figure 1.
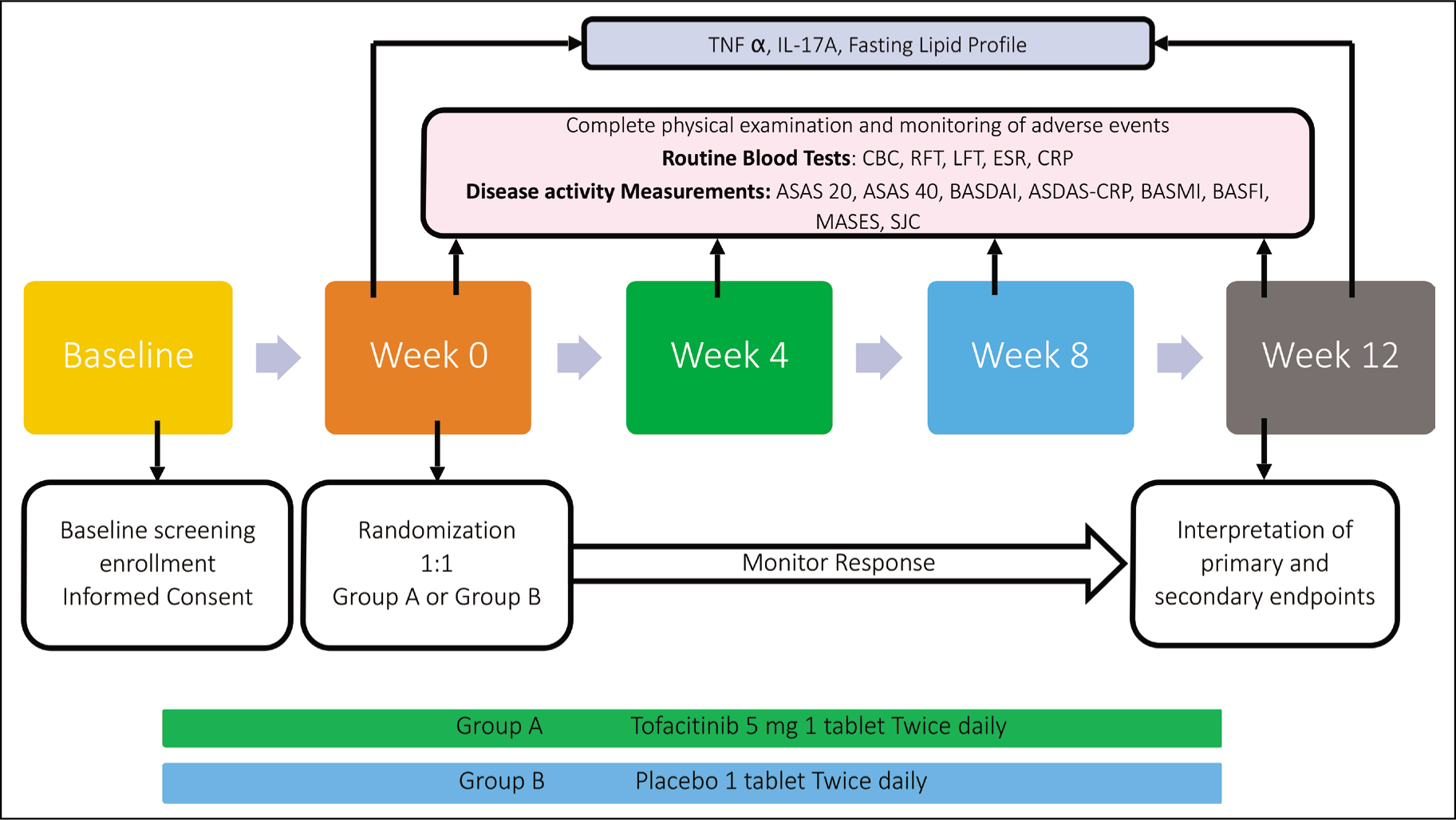
The randomised sequence was used to allocate tofacitinib or placebo to study participants and was blinded to the investigator by using SNOOSE. The drug was dispensed by technical staff, and the outcomes were assessed by the primary investigator.
Statistical Analysis
Microsoft Office Excel spreadsheet was used to enter the data. Statistical Package for the Social Sciences version 23.0 was used for statistical analysis. Qualitative variables were expressed as numbers and percentages, whereas quantitative variables were expressed as mean and standard deviation (SD) or median (interquartile range). Randomised patients who received the trial medication were all subjected to the complete analysis set and the safety analysis set. Intent-to-treat analysis was done. In contrast, only data during the on-drug period for study participants were used in the efficacy analyses. The change in disease activity and functional capacity parameters like ASDAS–CRP, BASDAI, BASMI, BASFI, SJC 44, MASES, inflammatory markers like ESR, CRP, serum TNF-α and IL-17 levels from baseline to week 12 were evaluated using the Wilcoxon rank-sum test. The mean change between the groups, tofacitinib and placebo, was evaluated using a two-independent samples test. A P value < .05 was considered a statistically significant value.
Results
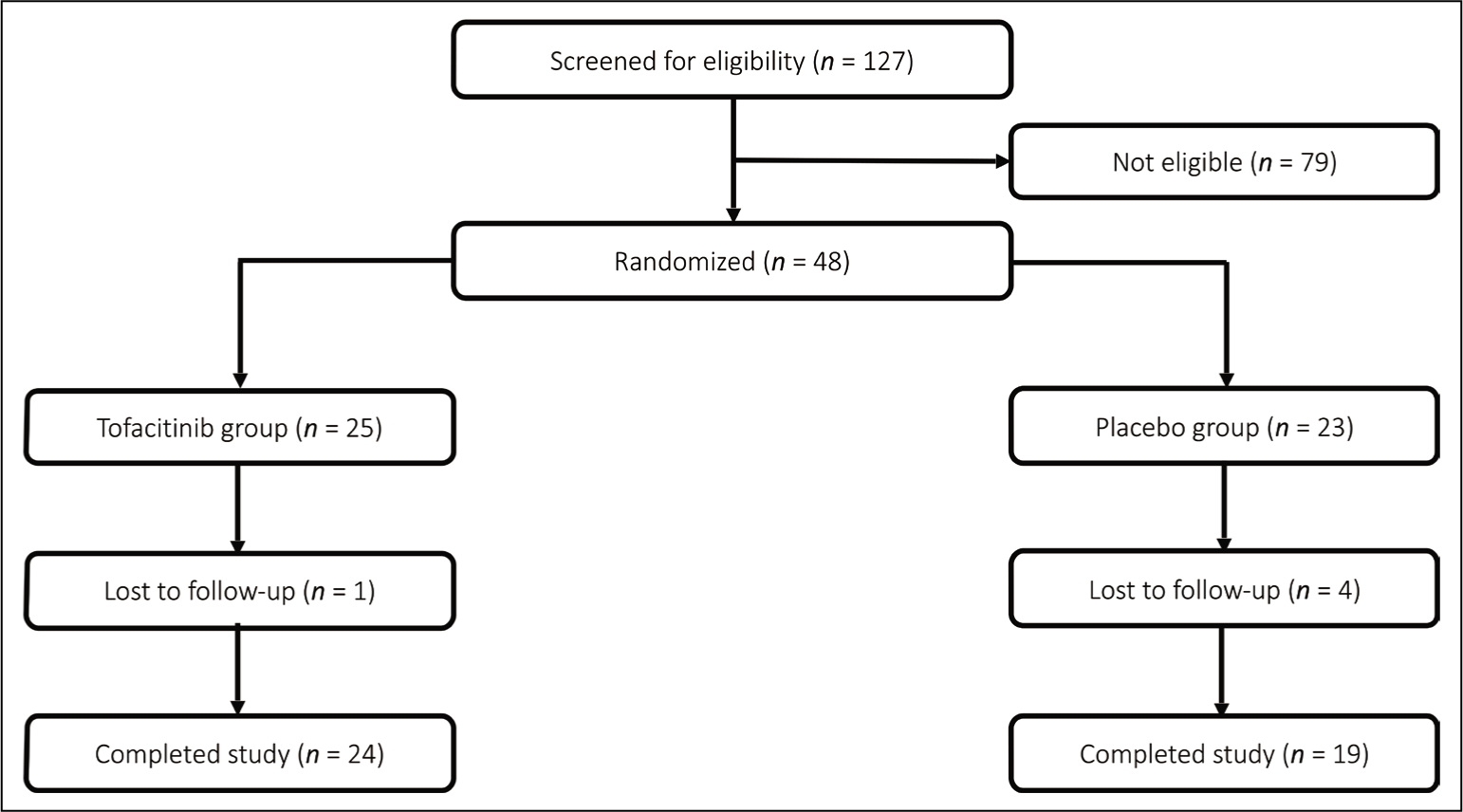
Of the total 127 patients with AS screened for eligibility, 48 patients fulfilled inclusion criteria and received either tofacitinib (25 patients) or placebo (23 patients). One patient in the tofacitinib group and four patients in the placebo group were lost to follow-up. Figure 2 shows patient disposition over the study period.
Patient Disposition Over the 12-week Study Period.
The majority of the study participants were male with a mean age of 36 ± 10.6 years and a mean disease duration of 4.5 years. The mean duration since diagnosis was 2.8 years, with 80% of patients between 2 and 5 years of disease duration. Peripheral arthritis was the most common extra-axial involvement (72%), followed by enthesitis (56%). Six patients had previously experienced uveitis episodes, whereas one patient in the tofacitinib group had acute iritis at the time of study inclusion. Three patients were diagnosed with inflammatory bowel disease, and none had psoriasis. All patients had bilateral sacroiliitis of grade 2 or more. Syndesmophytes were noted in 27% of patients, commonly affecting the thoracic spine (22%), followed by the lumbar and cervical spine. Three patients had hip involvement. The study cohort included active AS with a mean BASDAI of 5.4 and ASDAS–CRP of 4.2. The demographic and clinical characteristics of the study groups are shown in Table 1.
Demographics and Baseline Characteristics of Study Groups.
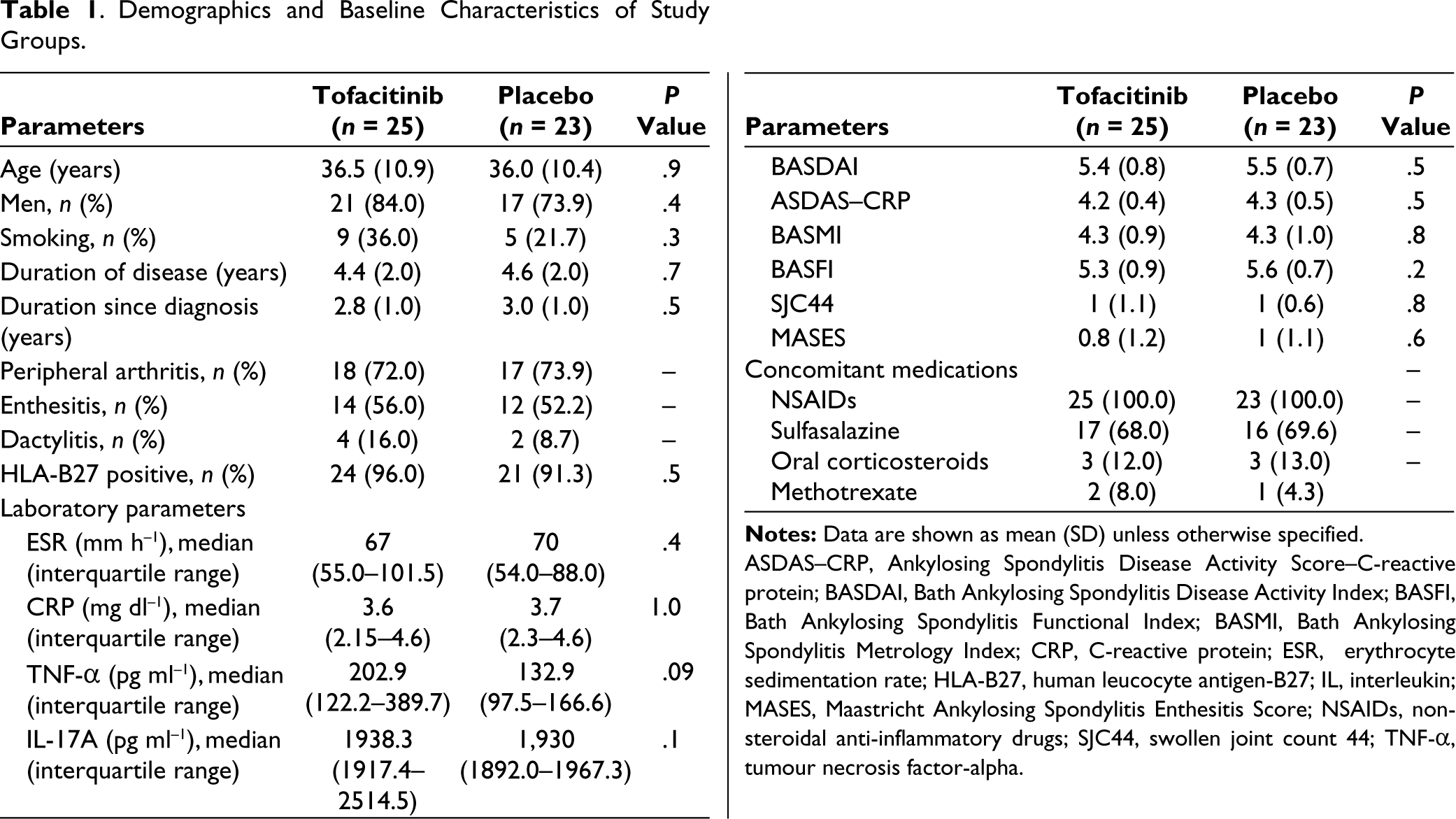
ASDAS–CRP, Ankylosing Spondylitis Disease Activity Score–C-reactive protein; BASDAI, Bath Ankylosing Spondylitis Disease Activity Index; BASFI, Bath Ankylosing Spondylitis Functional Index; BASMI, Bath Ankylosing Spondylitis Metrology Index; CRP, C-reactive protein; ESR, erythrocyte sedimentation rate; HLA-B27, human leucocyte antigen-B27; IL, interleukin; MASES, Maastricht Ankylosing Spondylitis Enthesitis Score; NSAIDs, non-steroidal anti-inflammatory drugs; SJC44, swollen joint count 44; TNF-α, tumour necrosis factor-alpha.
Endpoints
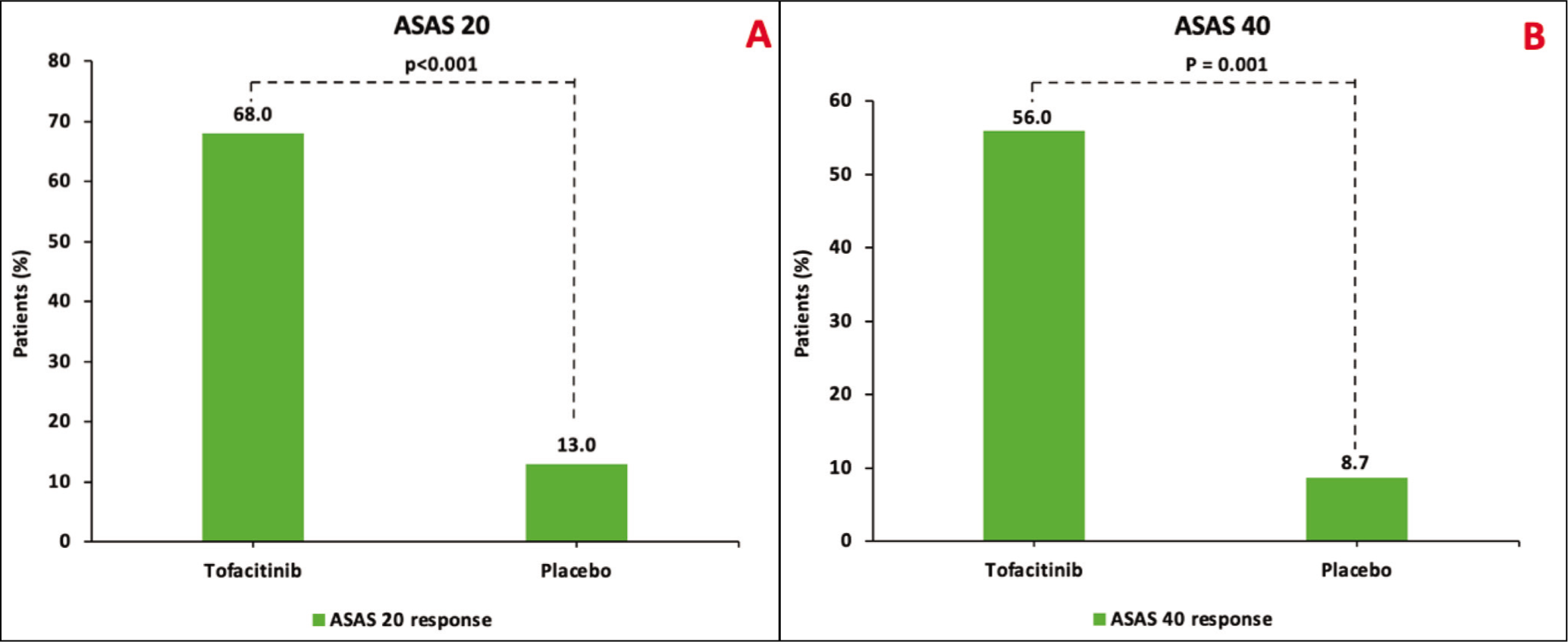
The tofacitinib group had higher response rates (ASAS20 and ASAS40) compared to the placebo group at 12 weeks (68.0% vs 13%, P < .001 and 56% vs 8.7%, P = .001, respectively; Figure 3A and B). Disease activity measures were significantly better (∆BASDAI [P < .001], ∆ASDAS–CRP [P <.001], ∆BASMI [P = .003], ∆BASFI [P = .003], ∆SJC44 [P = .017], ∆ESR [P = .002], ∆CRP [P = .003], ∆TNF-α [P = .007] and ∆IL-17A [P <.001]) in the tofacitinib group vs placebo group. However, ∆MASES (P = .76) was not different between the groups. The primary and secondary endpoints in both groups at 12 weeks are compared in Table 2.
Subgroup analysis showed significantly higher disease activity measures in non-responders to tofacitinib than responders (BASDAI: 5.8 vs 5.1, BASMI: 5.0 vs 3.9 and BASFI: 6.0 vs 4.9). The baseline characteristics of responders and non-responders in the tofacitinib group are compared in Table 3.
Safety
Infections were the common adverse effect, with upper respiratory tract infection (URTI) episodes numerically more in the tofacitinib group (n = 6) vs placebo group (n = 3). Five out of nine patients with URI required oral antibiotics, whereas the remaining patients received symptomatic treatment. All infections were non-severe and did not require hospitalisation. The occurrence of tinea infections was not different. The placebo group had one non-serious herpes zoster infection requiring anti-viral treatment. None of the patients had cytopenia, transaminitis, dyslipidaemia, severe infections or MACE throughout the study period. Table 4 summarises the adverse effects during the study period.
Comparison of Primary and Secondary Endpoint Measures Between Tofacitinib and Placebo Groups at Week 12.
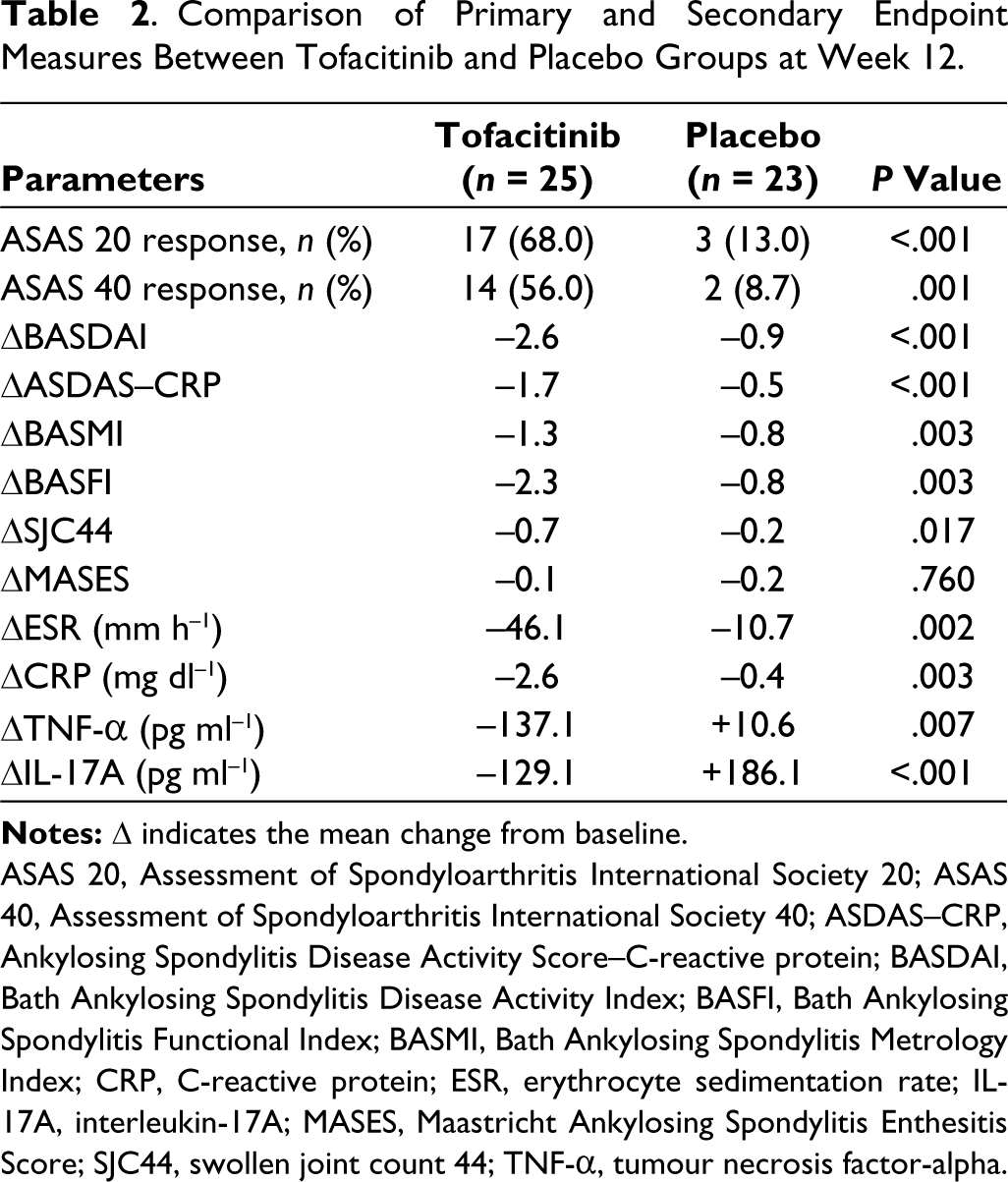
ASAS 20, Assessment of Spondyloarthritis International Society 20; ASAS 40, Assessment of Spondyloarthritis International Society 40; ASDAS–CRP, Ankylosing Spondylitis Disease Activity Score–C-reactive protein; BASDAI, Bath Ankylosing Spondylitis Disease Activity Index; BASFI, Bath Ankylosing Spondylitis Functional Index; BASMI, Bath Ankylosing Spondylitis Metrology Index; CRP, C-reactive protein; ESR, erythrocyte sedimentation rate; IL-17A, interleukin-17A; MASES, Maastricht Ankylosing Spondylitis Enthesitis Score; SJC44, swollen joint count 44; TNF-α, tumour necrosis factor-alpha.
Comparison of Baseline Characteristics in Responders and Non-responders in the Tofacitinib Group.
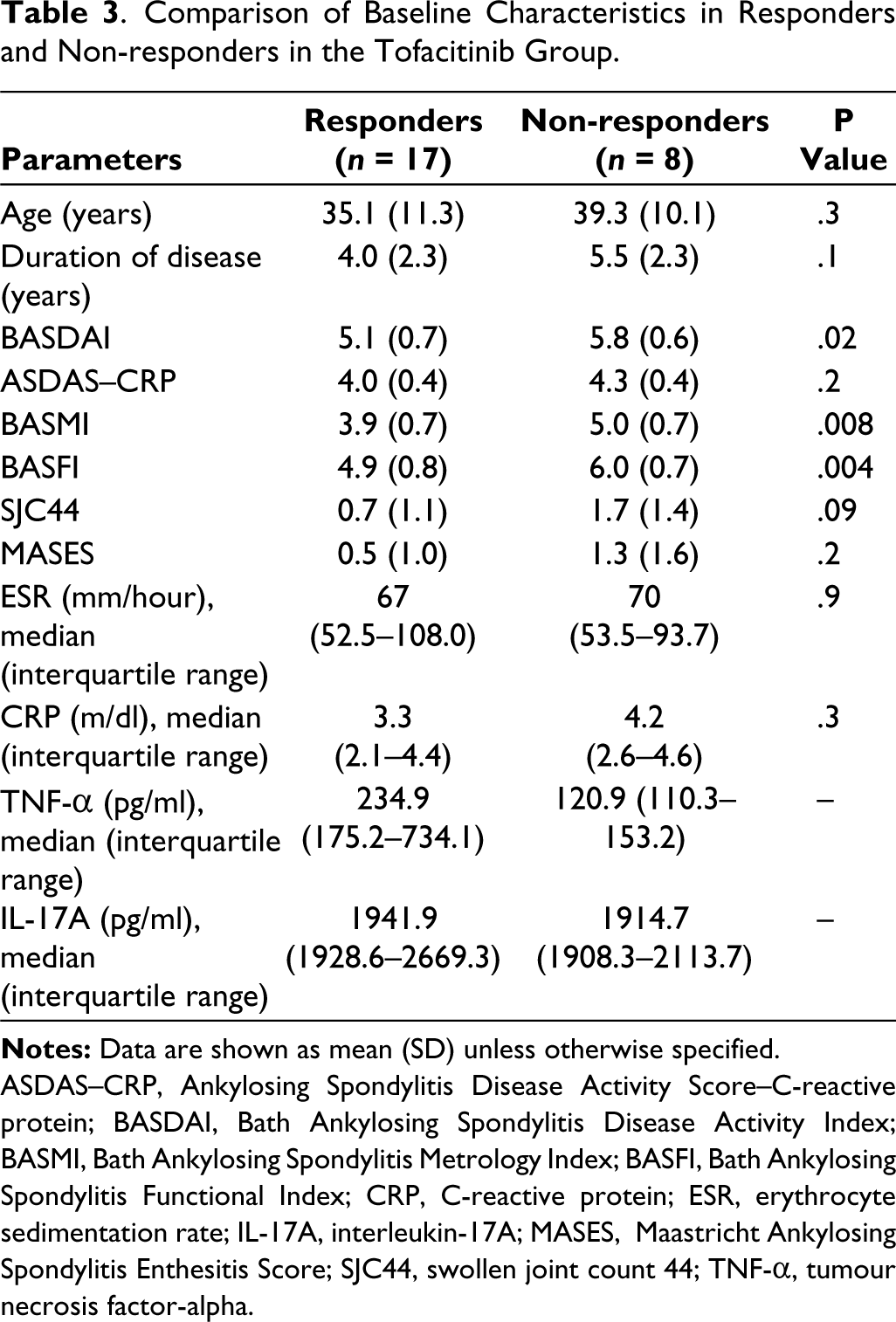
ASDAS–CRP, Ankylosing Spondylitis Disease Activity Score–C-reactive protein; BASDAI, Bath Ankylosing Spondylitis Disease Activity Index; BASMI, Bath Ankylosing Spondylitis Metrology Index; BASFI, Bath Ankylosing Spondylitis Functional Index; CRP, C-reactive protein; ESR, erythrocyte sedimentation rate; IL-17A, interleukin-17A; MASES, Maastricht Ankylosing Spondylitis Enthesitis Score; SJC44, swollen joint count 44; TNF-α, tumour necrosis factor-alpha.
Adverse Effects During the Study Period.
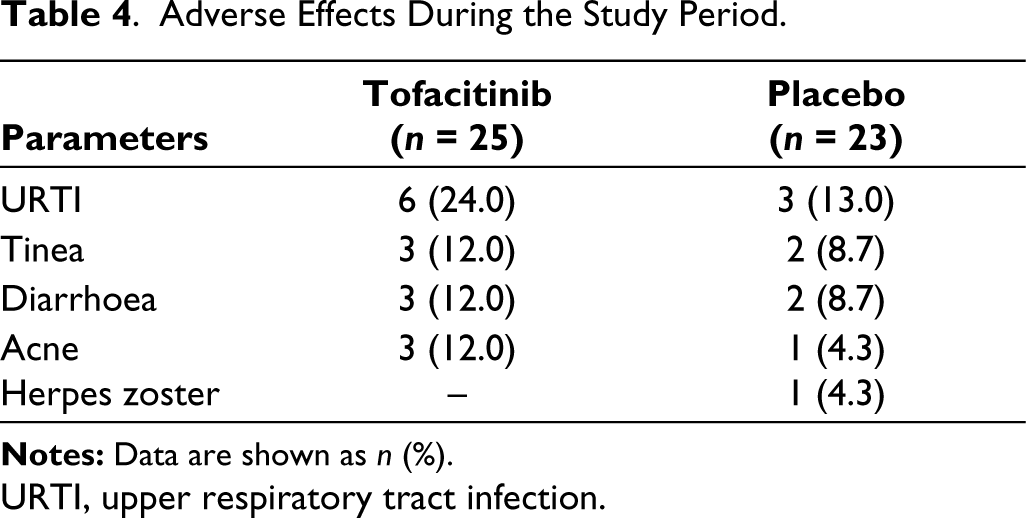
URTI, upper respiratory tract infection.
Discussion
This randomised control trial demonstrated the efficacy of tofacitinib over a period of 12 weeks in patients with AS. Tofacitinib given at a dose of 10 mg day−1 in divided doses in patients with active disease showed significant response, as evident in ASAS20 and ASAS40 response rates.
The efficacy of tofacitinib in AS was studied in two randomised, placebo-controlled trials, phase II and phase III by van der Heijde et al. 12 and Deodhar et al., 13 respectively. The phase II dose-ranging study showed significant improvement in ASAS 20 response with 5 mg twice daily dose compared to placebo (80.8% vs 41.2%, P ≤ .001). However, a considerable response was not achieved with 2 mg or 10 mg twice daily doses of tofacitinib. The ASAS40 response was markedly improved with all three doses of tofacitinib. 12 The results of the phase III trial showed a significant response with tofacitinib 10 mg day−1 in divided doses compared to placebo (ASAS20 response: 56.4% vs 29.4%, P < .0001; ASAS40 response: 40.6% vs 12.5%, P < .0001). 13 The clinical characteristics of patients in previously published studies on tofacitinib in AS and the present study are shown in Table 5.
Comparison of Clinical Characteristics of Patients in Previously Published Studies on Tofacitinib in AS and Present Study.
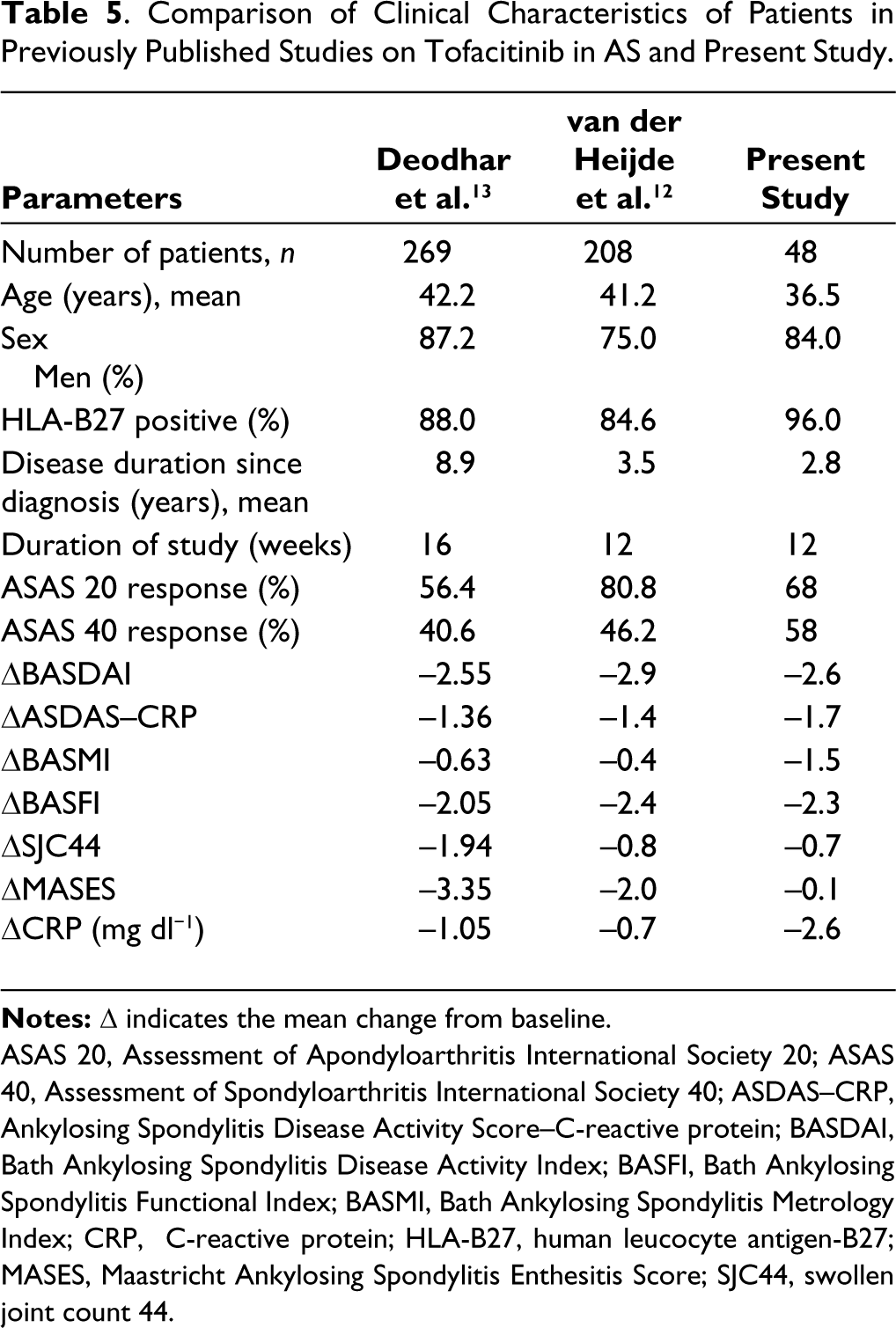
ASAS 20, Assessment of Apondyloarthritis International Society 20; ASAS 40, Assessment of Spondyloarthritis International Society 40; ASDAS–CRP, Ankylosing Spondylitis Disease Activity Score–C-reactive protein; BASDAI, Bath Ankylosing Spondylitis Disease Activity Index; BASFI, Bath Ankylosing Spondylitis Functional Index; BASMI, Bath Ankylosing Spondylitis Metrology Index; CRP, C-reactive protein; HLA-B27, human leucocyte antigen-B27; MASES, Maastricht Ankylosing Spondylitis Enthesitis Score; SJC44, swollen joint count 44.
The secondary endpoints, including disease activity and functional measures (BASDAI, ASDAS–CRP, BASMI and BASFI) and inflammatory markers (ESR and CRP), showed significant reduction with tofacitinib when compared to placebo. A similar marked improvement in BASDAI, ASDAS, BASMI, BASFI and CRP was noted in both phase II and phase III trials of tofacitinib in patients with AS.12,13
The subjects in our study cohort showed notable improvement in SJC44 in contrast to van der Heijde et al. 12 and Deodhar et al. 13 This was attributed to relatively fewer study patients at entry with swollen joints in the phase III study (Tofacitinib, 24.8% vs placebo, 27.9%), whereas our study had a higher number (11 patients in tofacitinib group, 44% vs 13 patients in placebo group, 56.5%). 13 Six patients in the tofacitinib group and one patient in the placebo group showed complete improvement by the end of 12 weeks.
The enthesitis score and MASES did not show significant improvement in this study. Contrasting results from phase II and phase III studies were seen, with the former showing a significant improvement in MASES as compared to placebo for both the 10 mg day−1 and 20 mg day−1 (both in divided doses), whereas the latter showed no difference for the 10 mg day−1 (divided dose) group.
Serum TNF-α and IL-17A were reduced at 12 weeks in patients on tofacitinib. The in vitro culture of CD4+ T cells under T helper 17 (Th17) cell-promoting conditions from patients with AS was done by Hammitzsch et al. 18 to demonstrate the therapeutic potential of four different JAK inhibitors, including tofacitinib. They observed that JAK inhibitors had significantly reduced the production of multiple Th17 cytokines including IL-17A, IL-17F and IL-22 by inhibiting CD4+ T cells. 18 Patients with rheumatoid arthritis on tofacitinib were studied by Li et al. 19 in order to quantify the drug’s effects on serum cytokine levels. Tofacitinib was associated with a notable, early (<4 weeks) reduction of disease activity as well as levels of serum TNF- α, IL-17, IL-6 and IFN-γ. Fitz et al. 20 evaluated the efficacy of tofacitinib and etanercept in psoriasis and correlated them with serum IL-17A levels. There was a reduction in IL-17 levels in responders.
In the subgroup analysis, non-responders had higher disease and functional activity indices, including BASDAI, BASMI and BASFI, at baseline. Further prospective studies of tofacitinib are required to determine the factors that could predict the selection of patients and favourable outcomes for tofacitinib in AS.
The common adverse effects reported were URTIs and diarrhoea, which was similar to the study by Deodhar et al. 13 In addition, taenia infection and acne were also reported. Both patient groups receiving tofacitinib or placebo showed a similar frequency of infections. No major cardiovascular events or serious detrimental effects, thromboembolic events, cytopenia, dyslipidaemia, transaminitis or herpes zoster were observed in patients who received tofacitinib.
Strengths
The strength of the study includes its randomised design, ensuring unbiased allocation of participants to treatment groups. Additionally, the placebo-controlled design enables precise comparison between the study groups, enhancing validity and reliability and providing better insights into the efficacy and safety of tofacitinib.
Limitations
Our study had several limitations. First, the sample size was small. The limited sample size in each group made it challenging to understand potentially intriguing subgroup analyses. Second, the shorter follow-up period of 12 weeks did not allow for assessing the long-term effects of tofacitinib therapy. The 12-week duration of the study was considered after taking into account the benefits of using tofacitinib in previous clinical trials, maintaining a balance between patient safety and limited exposure time. There is a possibility that the duration of the study was insufficient to achieve the maximal efficacy of the drug. Furthermore, due to the short follow-up period, radiographic progression was not studied.
Conclusion
In patients with active AS on tofacitinib, a greater reduction of disease activity measures was noted versus placebo and significantly reduced levels of TNF-α and IL-17A without any new concerning safety risks.
Footnotes
Authors’ Contributions
Pradeepta Sekhar Patro and BV Harish were involved in the concept and design of the study. BV Harish was involved in data acquisition. Rasmi Ranjan Sahoo, Samanyoya Gochhait and BV Harish analysed the data and drafted the manuscript. Pradeepta Sekhar Patro and Rasmi Ranjan Sahoo supervised the entire work. All authors critically revised the manuscript and approved the final draft.
Code Availability
Not applicable.
Consent for Publication
All authors consent to publication and grant the publisher exclusive licence of the full copyright.
Consent to Participate
All study participants signed informed consent before enrolment.
Data Availability
The authors confirm that the data supporting the findings of this study are available within the article and/or its supplementary materials. Further clarifications as required will be available from the corresponding author, Pradeepta Sekhar Patro, upon reasonable request.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Ethical Approval
The study was approved by the institutional ethics committee of IMS & SUM Hospital, Bhubaneswar, India (ref no.: IEC/IMS.SH/SOA/2021/275).
Funding
The authors received no financial support for the research, authorship and/or publication of this article.