
Abstract
Systemic juvenile idiopathic arthritis (SJIA), presently included under the broad taxonomy of Still’s disease along with ‘Adult-onset Stills disease’, is a unique disease entity, characterised predominantly by features of systemic inflammation at disease onset and running a heterogenous course, evolving into a disabling chronic, destructive arthritis in a sizable proportion of patients. With a better understanding of the disease biology, there has been a paradigm shift in therapeutic strategy targeting immunological remission. With the evolution of better biomarkers (to guide diagnosis, predict complications and predict treatment responses) and robust therapeutic options and treatment targets, newer challenges such as refractory disease states and SJIA-associated lung disease have risen to the fore, making disease management all the more challenging.
Keywords
Introduction
Systemic juvenile idiopathic arthritis (SJIA) is a distinctive juvenile febrile illness with its immunopathology at the crossroads between auto-inflammation and auto-immunity. 1 Although considered a type of juvenile idiopathic arthritis by the International League of Associations for Rheumatology (ILAR), SJIA stands out among the rest by way of its complex immunopathology, natural history, and response to treatment. 2
With the advent of better insight into disease biology and a paradigm shift in therapeutic approach from predominantly glucocorticoid-based step-up therapy to upfront targeted biologic therapy, newer challenges such as SJIA-lung disease and refractory SJIA are being unravelled, exciting more questions with every answer discovered.
This review attempts to elaborate on the natural history, immunopathology, biomarkers, therapeutic landscape, and challenges in the diagnosis and management of SJIA.
Epidemiology
JIA is the most prevalent rheumatic disease of childhood with an estimated incidence of 1.6 to 23/ Lakh and a prevalence of 3.8–400/lakh population. 3 SJIA comprises 10%–20% of JIA with an incidence of 0.4–0.8/Lakh. 4 and pooled prevalence estimate of 0–8.6/Lakh. 3 and commonly affects children between 1 and 5 years of age with no sex predilection. 5
Greater heterogeneity is noted in JIA phenotype within the Indian population, with SJIA being more prevalent down south comprising close to 32% of JIA 6 in contrast to around 27% in North India, where ERA JIA predominates 7 ; 20% in Eastern India 8 and close to 30% in western India, 9 where Polyarticular JIA predominates. Whether this represents a heterogenous genetic landscape or varied environmental exposure or just a referral bias remains debatable.
Clinical Features
SJIA is one of the most important differential diagnoses in a child presenting with pyrexia of unknown origin. It is prudent to suspect SJIA in any child presenting with an unaccounted prolonged febrile episode with a typical pattern and associated symptoms as described below.
Fever
SJIA is characterised by a distinctive pattern of fever, characterised by temperature spikes crossing 39°C once or twice a day with rapid return to normalcy within a few hours. The peaks of fever can make the child profoundly ill-looking, albeit contrasting normalcy in the inter-febrile period. 10 However, the classic pattern of fever is observed only in around 37% of children. 5 The evolution of intermittent fever patterns to continuous high-grade fever usually suggests the onset of macrophage activation syndrome (MAS). 11 Also, MAS can be the presenting manifestation in 22%–53% of children with SJIA.12,13
Rash
Evanescent, asymptomatic, erythematous macular or maculopapular rash distributed over trunk, neck and proximal extremities, is observed during peaks of temperature in more than 80% of children and resolves completely without residual scarring or pigmentation at fever defervescence.5,14 Hence, the rash may be easily missed in a dark-skinned child or if not observed, during the peak of fever. Other less common cutaneous lesions include migratory non pruritic erythema displaying Koebner phenomenon 15 and urticarial lesions. 16
Articular Manifestations
Although arthritis is a mandate for meeting ILAR classification criteria for SJIA, arthralgia alone is commonly found at disease onset, and true arthritis evolves within weeks, months, or rarely years later (most commonly within 3 months of disease onset). 10
The pattern of arthritis may be variable, ranging from migratory oligoarticular to fixed polyarticular patterns. The most commonly involved joints include the wrist, knee, and ankle. 5 The hip, 17 cervical spine, 18 temporomandibular joint, 19 and small joints of the hands and feet 17 are the other commonly affected joints. Without early, appropriate treatment, arthritis can be rapidly destructive (radiographic evidence of joint space narrowing is noticed as early as 2 years after disease onset), resulting in joint space loss, erosions, and ankylosis. 20 Synovial cysts are known to occur in SJIA due to persistent articular inflammation and the resultant tracking of inflammatory fluid along the tendon sheath. 21 They can present as sudden-onset swelling or rupture to present as pseudo-thrombophlebitis. 22 The most common site observed is the brachial synovial cyst communicating with the shoulder joint. 23 Tenosynovitis and myalgia are other common musculoskeletal manifestations.
Serositis
Although serositis is mostly asymptomatic and is incidentally detected on imaging, acute pericarditis can be symptomatic causing chest pain and can evolve rapidly into cardiac tamponade. 10 Pleuritis is also observed less frequently.
Reticuloendothelial Manifestations
Generalised lymphadenopathy (in over 25% of children) and hepatosplenomegaly are less frequently observed during the active phase of the disease.24,5
Other Manifestations
Other less common manifestations include a sore throat, aseptic meningitis, 25 pulmonary alveolar proteinosis, interstitial lung disease and pulmonary hypertension. 26
Serial monitoring of blood counts, inflammatory markers, serum ferritin, liver enzymes, and coagulation profile, rather than one-time evaluation at disease onset, is crucial to guiding therapy. The laboratory and radiological features of SJIA are illustrated in Table 1 and Table 2 respectively. The clinical, laboratory and radiographic features are summarized in Figure 1.
Laboratory Features of SJIA.

Radiological Features of SJIA.
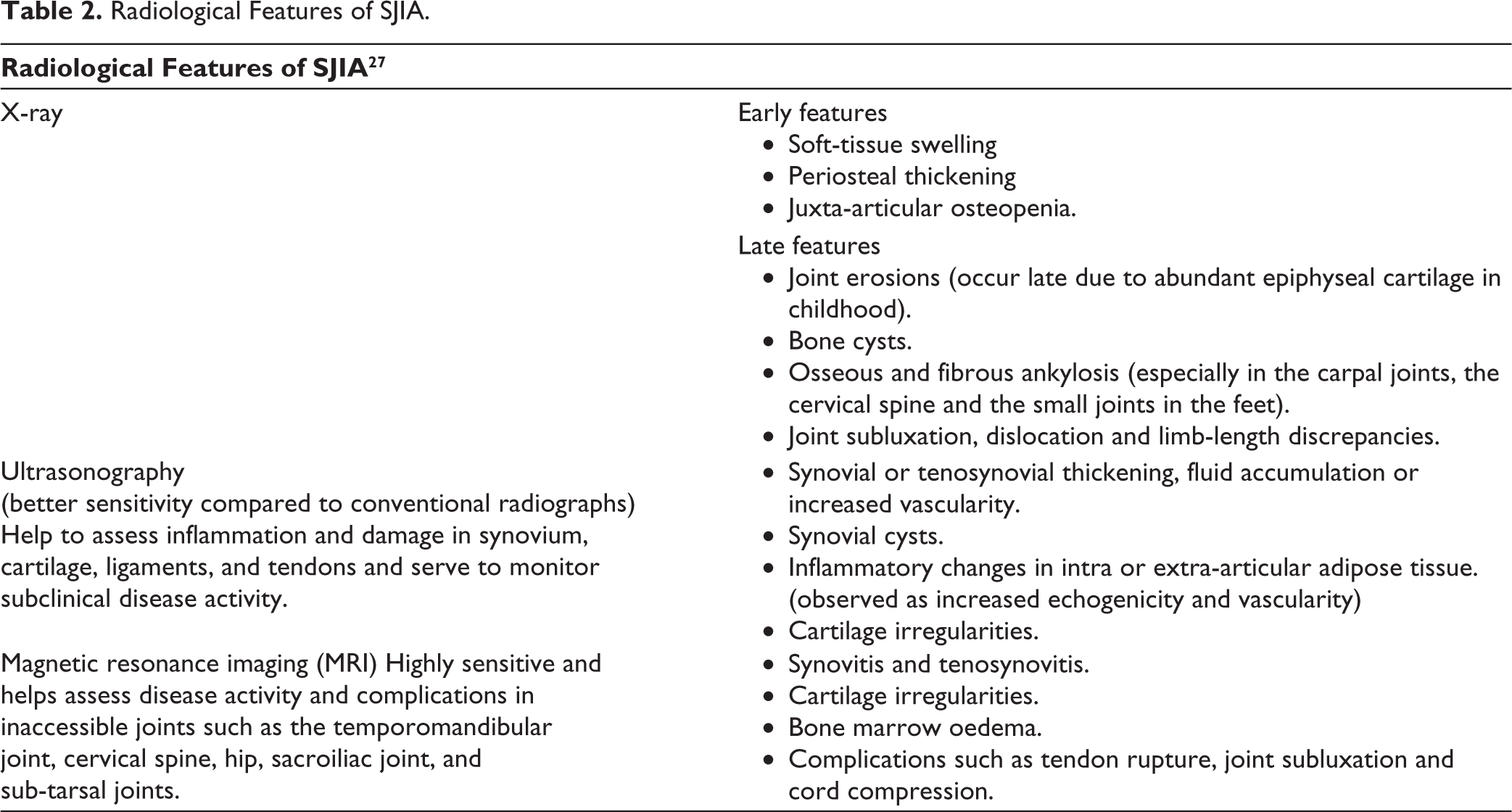
Summary of Clinical and Laboratory Features of SJIA.
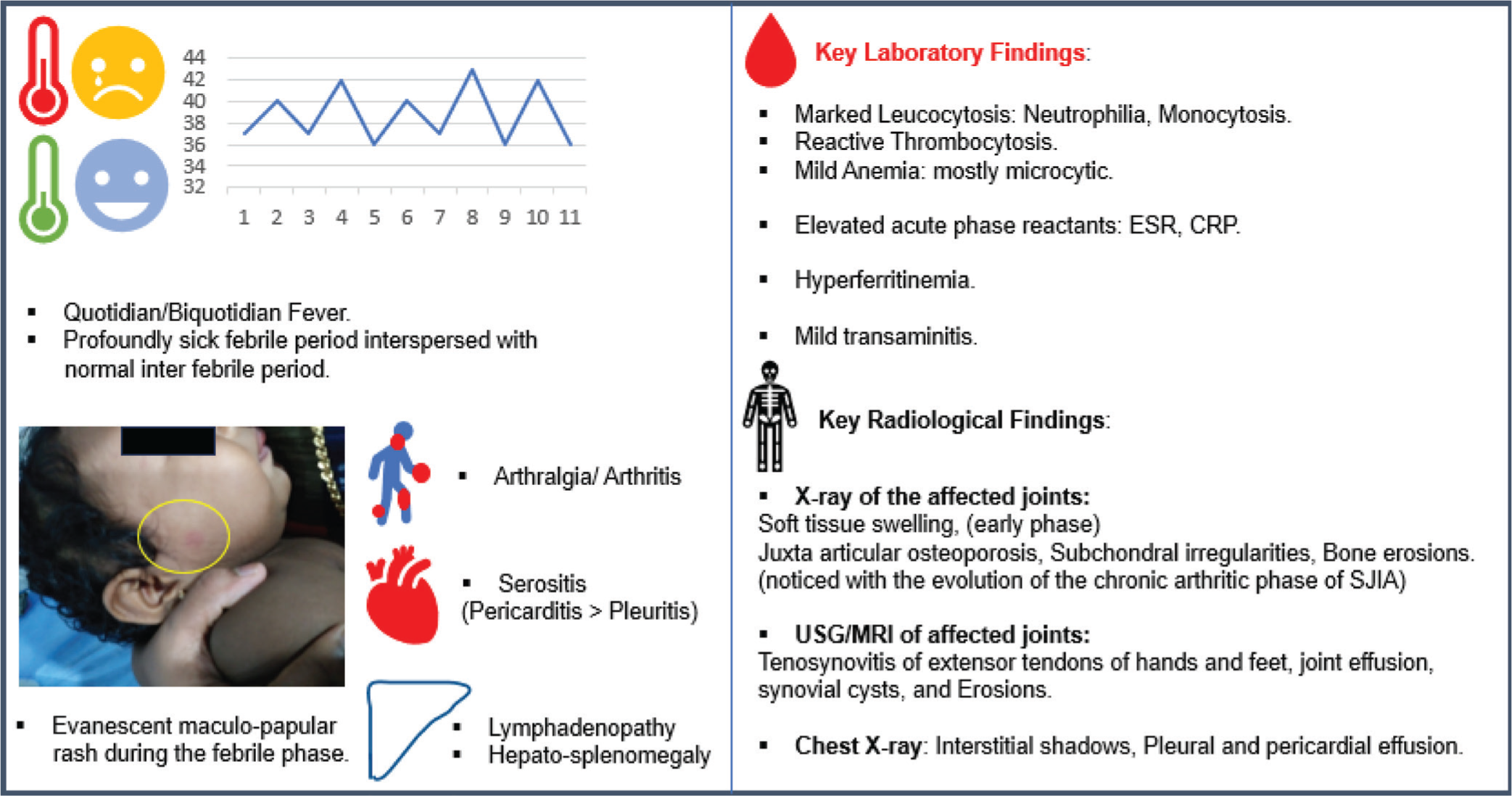
Etiopathogenesis
SJIA is characterised by prominent systemic symptoms at disease onset mimicking a monogenic hereditary fever syndrome followed by progression to chronic destructive arthritis suggesting the possibility of a biphasic disease model with a window of opportunity.
Genetic Risk Factors
MHC class II association is the strongest predicted genetic risk factor for disease onset, especially in children. HLA DRB1*11 is known to be associated with SJIA in the Asian population. 1
LACC1 gene polymorphism (Laccase domain containing protein-1) resulting in LACC-1 dysfunction is a known cause of the monogenic recessive SJIA phenotype. The LACC1 gene encodes the protein FAMIN, which regulates the metabolism of macrophages and inflammasome activation. 28
IL-1RN gene polymorphism: IL-1 receptor antagonist (IL-1RN) is a key negative regulator of pro-inflammatory IL-1 signalling. Based on studies on IL-1RN knockout mice, it is found that hypomorphic IL-IRN polymorphisms, which cause lower levels of IL-1RN, are linked to a greater risk of SJIA. Also, it is observed that SJIA patients with higher expression levels of IL-1RN respond poorly to anakinra (an IL-1 receptor antagonist), although the association is not consistent in different populations. 29
Other postulated genetic defects include heterozygous TNFAIP3 and NLRC4 gene polymorphisms, impaired regulatory IL-10 activity and polymorphisms in VEGFC (vascular endothelial growth factor C), IL-18, MMIF (macrophage migration inhibitory factor), LILRA3 (leucocyte immunoglobulin-like receptor A3), CASP-1 (caspase-1), and PRF-1 (perforin 1) genes. 29
Nigrovic’s Biphasic Model of Disease Pathogenesis 30
Yet unidentified, pathogen-associated molecular patterns (PAMPS) and danger-associated molecular patterns (DAMPS) signal through pattern recognition receptors (PRRs), chiefly TLR-4,9 and NLRP-3, in a genetically predisposed individual and result in spontaneous, inappropriate, and uncontrolled activation of predominantly myeloid cells (Neutrophils >> Monocytes) during disease initiation, resulting in inflammasome activation and release of proinflammatory cytokines IL-1β, IL-18, alarmins (S100 A8/A9; S100 A12) and advanced glycation end products. Autocrine and paracrine IL-1 signalling results in downstream IL-6 generation, whose serum levels correlate with systemic symptoms.
This cytokine milieu, enriched with IL-1, IL-18, and IL-6, further shapes the adaptive cell repertoire by preferentially skewing T-helper cell polarisation from the Treg phenotype to the IL-17-secreting Th17 phenotype and inflammatory follicular (Tfh) and peripheral follicular T-helper cells (Tph) phenotype, driving B cell plasmablast generation.
Increased IL-18 production promotes T-cell proliferation, including NK T cell and γδ T cells. However, IL-18 resistant state is observed in SJIA due to defect in IL-18R (IL-18 receptor) and enhanced production of IL-18 binding protein (IL-18 BP) which leads to IFN-γ deficient state. This cytokine milieu of the early phase of SJIA favours T-cell polarisation to enhance IL-17-generating Th17 and Tfh/ph phenotypes, with reduced Treg and Breg cells and regulatory cytokines like IL-10. 31
The evolution of the immune phenotype from the predominant pro-inflammatory myeloid phenotype characterised by high IL-1β, IL-6, and IL-18, to the pro-inflammatory adaptive phenotype, characterised by enrichment of IL-17-secreting Th17, Tfh/Tph, NK T cells, and γδ T cells, paves the way for the evolution of the clinical phenotype of SJIA from predominant systemic inflammation mimicking autoinflammatory syndromes to chronic destructive arthritis. The etiopathogenesis of SJIA and its complications are as illustrated in Figure 2.
Etiopathogenesis of SJIA and MAS.19,18,20
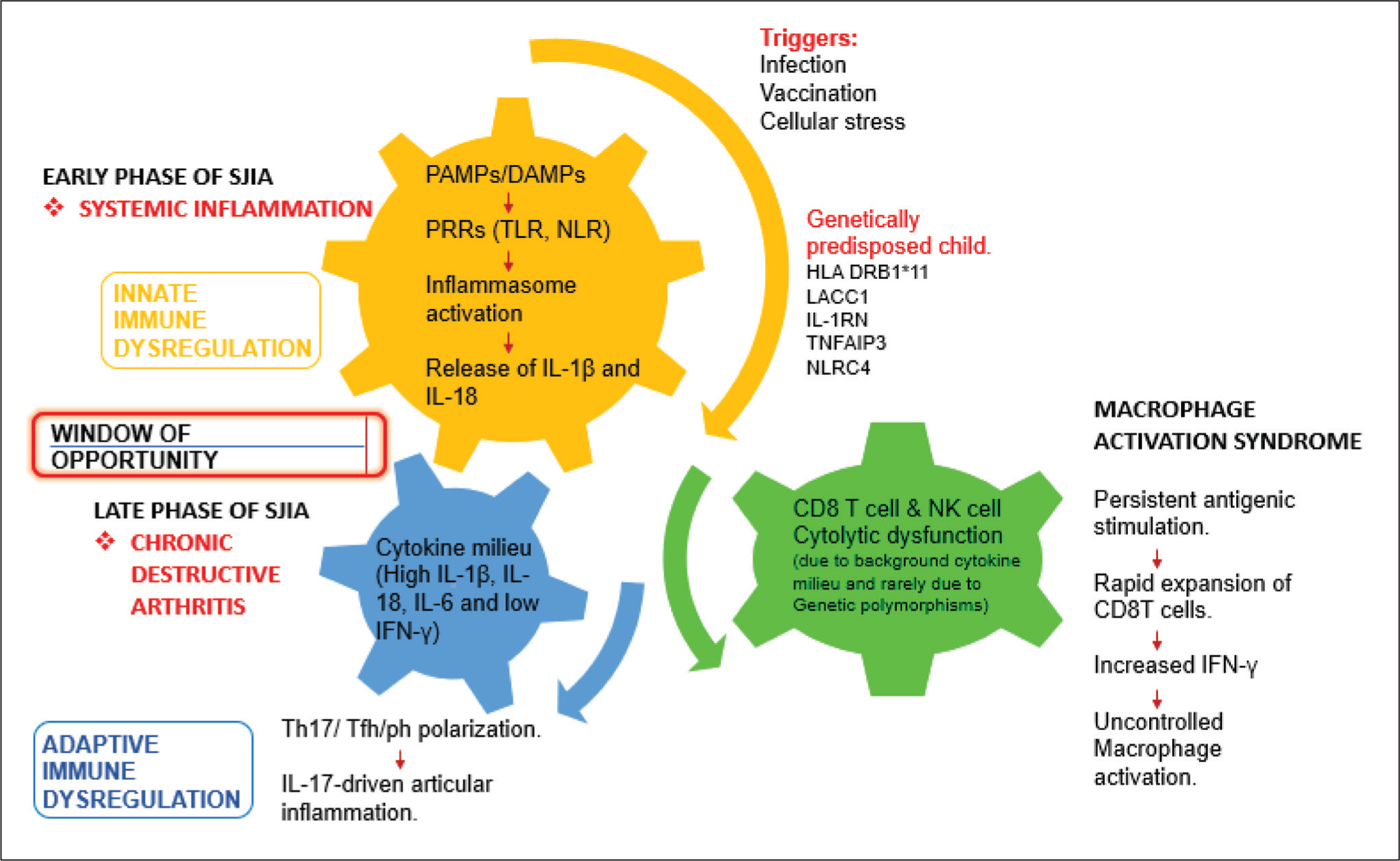
This insight about evolution of the proinflammatory immune landscape from a predominant myeloid signature to subsequent IL-17 predominant adaptive signature, led to the concept of the ‘window of opportunity’, hypothesising that effective control of systemic inflammation at disease onset using appropriate biologic therapy targeting IL-1 or IL-6 can prevent the development of a pathological adaptive effector phenotype, thereby achieving immunological remission and preventing crippling morbidity associated with the evolution of destructive arthritis. 32
Natural History of SJIA
The disease course in SJIA is heterogeneous, and currently, there are no defined clinical or lab findings that predict the trajectory of SJIA at an individual level. It is observed from various cohorts that around 40% of children with SJIA follow a monophasic course and achieve complete remission after a few months of disease activity (typically lasting for <24 months); 7% follow a polyphasic course characterised by recurrent disease flares interspersed with months or years of remission; and surprisingly, a major proportion (nearly 50%) follow a persistent course lasting for at least 2 years. 33 Polyarticular presentation at onset and evidence of ongoing disease activity at 3 months are observed as risk factors predicting a persistent disease course. The natural history of SJIA is as illustrated in Figure 3. 34
Natural History of SJIA – 40% Children Follow Monophasic Course, 7% Polyphasic and Close to 50% Chronic Persistent Disease Course.
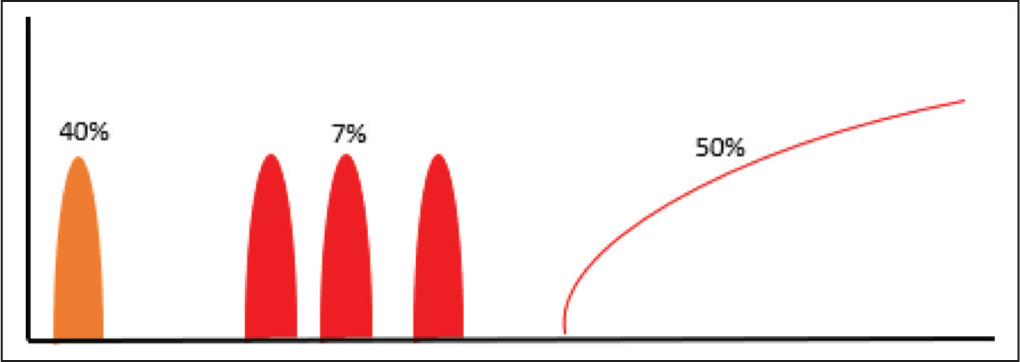
Clinical and Immunological Clusters
With the understanding that SJIA and Adult-onset Stills disease (AOSD) lie along the continuous spectrum of a single disease (‘Stills disease’), different clinical clusters are being recognised, which may help predict the disease course and aid therapeutic stratification. The proposed clinical clusters include juvenile/transitional, uncomplicated, hyperferritinemic and catastrophic. 35
Complications
Refractory SJIA
Despite the widespread use of upfront biologics, various longitudinal follow-up cohort studies such as Nordic 36 and ReACCh-Out 37 have shown that a considerable proportion of patients with SJIA are refractory to treatment, including biologics and need sequential trials of multiple biologics and combination therapies, and can become steroid dependent for adequate disease control. Although there are no definite validated criteria for labelling SJIA as refractory, three different disease courses are considered to typify refractory SJIA, which are as illustrated in Table 3. 34
Course of Refractory SJIA.

Macrophage Activation Syndrome (MAS)
MAS is a potentially fatal complication of SJIA that can be triggered by infections such as EBV, CMV, and influenza or by persistent, uncontrolled SJIA disease activity. Although clinically overt MAS is observed in 10%–15% of children with SJIA, subclinical MAS is observed in as many as 30%–40% of children. MAS can be the presenting manifestation in 22%–53% of children with SJIA. 34
Persistent, inappropriate signalling through PRRs (pattern recognition receptors) and elevated pro-inflammatory cytokines IL-1β, IL-18 and IL-6, combined with cytolytic defects in T cells and NK cells, result in failure to clear the inciting immune trigger and result in persistent antigenic stimulation of macrophages, rapid proliferation of CD8 T cells, and massive production of IFN-γ, which contribute to a hyperferritinemic hyperinflammatory state termed MAS. 38
Clinically, the onset of MAS is characterised by a change in fever pattern from intermittent to continuous, unremitting fever; evanescent asymptomatic rash to fixed pruritic rash; the development of purpura and bruising with the onset of coagulopathy; more pronounced and generalised lymphadenopathy; hepatosplenomegaly; acute encephalopathy; and multi-organ failure if left untreated. The laboratory features of MAS are as illustrated in Table 4.
Laboratory Features of Macrophage Activation Syndrome.

Ravelli’s classification criteria 11 is used for distinguishing SJIA-associated MAS from infections or active SJIA and is largely rooted in the presence of hyperferritinemia. Despite this being non-specific, extreme hyperferritinemia (serum levels > 10,000 ug/L) indeed points towards overt or rapidly evolving MAS. The classification criteria for Macrophage activation syndrome is as illustrated in Table 5.
2016 Classification Criteria for Macrophage Activation Syndrome. 11

MAS can evolve into multi-organ dysfunction and can be rapidly fatal, with a mortality rate of around 8%–12%. 34 No single sign, symptom, or lab test has been proven to be diagnostic. Careful observation of clinical trends as detailed earlier forms the key to early diagnosis.
SJIA – Lung Disease
With more widespread use of upfront biologics, SJIA lung disease is becoming increasingly recognised. Hypersensitivity DRESS-like reactions to biologic therapy, the cytokine plasticity hypothesis, and the HLA Class II association, 39 incomplete suppression of pro-inflammatory cascades; none conclusive, have been varyingly put forward to explain the increased incidence of SJIA-LD. The risk factors for SJIA-LD are as illustrated in Table 6.
Risk Factors for SJIA – Lung Disease. 40

Lung involvement is usually seen early in the disease course (within 2 years of SJIA disease onset) and manifests clinically as resting tachypnoea, mild dyspnoea, and characteristic acute erythematous digital clubbing. 40 It is imperative to note that, usually, there is a striking dissociation between clinical signs and the extent of radiological lung involvement. The radiological features of SJIA - Lung disease are as illustrated in Table 7.
Radiological Features of SJIA. 40

Lung biopsy and histopathological examination show an overlapping endogenous lipoid pneumonia-pulmonary alveolar proteinosis (PAP) pattern. It differs from primary PAP by way of predominant lymphoplasmacytic inflammation, fibrosis with lobular remodelling and architectural distortion and scant eosinophilic proteinaceous material and lipid-laden macrophages.
BAL fluid analysis usually contains minimal eosinophilic proteinaceous material and is usually non-informative for the diagnosis of SJIA-LD. However, BAL may be helpful in ruling out infections.
SJIA-LD can have three different trajectories. 50% remain stable, 25% improve, and 25% progress over 1 year, and it is associated with a mortality rate as high as 37%. Hence, a high index of suspicion in at-risk children and a thorough evaluation with a 6-minute walk test, diffusion capacity of the lung for carbon monoxide, and a low threshold for imaging with HRCT is essential for timely diagnosis and prompt initiation of treatment. 40
Uveitis
Although uveitis is most commonly found in early-onset, ANA-positive, oligoarticular JIA subsets, less than 2% of children with SJIA can develop uveitis, and thus annual uveitis screening is recommended. 41 Regular ophthalmologic examinations are also helpful in monitoring steroid-induced side effects such as cataracts and glaucoma.
Amyloidosis
Secondary amyloidosis is a long-term complication of uncontrolled SJIA disease activity. It was an important cause of mortality before the advent of effective anti-inflammatory therapies. 10
Treatment
With a better understanding of the immunobiology of SJIA, a paradigm shift is observed in therapeutic strategy from a glucocorticoid-based step-up approach to treat to target approach with use of upfront targeted cytokine blockade.
Treatment of SJIA
Step-up Approach
The CARRA (Childhood Arthritis and Rheumatology Research Alliance) consensus treatment plan 2012 provides four different strategies, including glucocorticoid treatment plans, methotrexate, anakinra, and tocilizumab treatment plans, which may be combined with glucocorticoids. 42
This involves the initiation of treatment with either oral prednisolone 1–2 mg/kg/day upfront or following the initial 3–5 days of treatment with IV methylprednisolone 30 mg/kg/day. This is followed by reassessment at 1 month and 3 months and steroid tapering as permitted by disease activity. If the patient continues to require ≥50% of the starting steroid dose for controlling disease activity at 3 months of follow-up, second-line immunosuppression is introduced with oral or subcutaneous methotrexate, which is initiated at a dose of 0.5 mg/kg/week up to a maximum dose of 1 mg/kg/week (doses above 25 mg/week have no added benefit). Despite the addition of methotrexate, if the patient continues to require ≥50% of the starting steroid dose at 3 months of follow-up, biologic anti-IL-1 (anakinra) is initiated at a dose of 2 mg/kg subcutaneously daily up to a maximum dose of 4 mg/kg/day, or anti-IL-6 (Tocilizumab) is initiated at a dose of 8 mg/kg (if ≥30 kg) or 12 mg/kg (if ≤30 kg) intravenous every 2 weeks and switched to an alternate strategy in case of lack of response at 3 months. 42
Treat-to-Target Approach 43
With the advent of the concept of ‘window of opportunity’ in preventing evolution into refractory disease states, emphasis is being laid on early control of inflammation using upfront targeted therapies. Accordingly, the German PRO-KIND initiative and ACR 2021 guidelines recommend the upfront use of anti-IL-1 or anti-IL-6 therapy. Timely treatment targets are defined by the PRO-KIND initiative, and treatment is optimised accordingly.
This approach involves initiation of monotherapy with either anti-IL-1, anakinra (2 mg/kg/day subcutaneous to a maximum dose of 8 mg/kg/day) or canakinumab (4 mg/kg/4 weeks up to a maximum dose of 300 mg) or anti- IL-6, Tocilizumab (8 mg/kg intravenous every 2 weeks if ≥30 kg or 12 mg/kg every 2 weeks if ≤30 kg). Therapy is tailored with dose escalation of initial biologics, switch to alternate biologics or introduction of steroids if disease activity targets are not met. If arthritis alone persists after adequate control of systemic inflammation, methotrexate, leflunomide, anti-TNF or abatacept can be tried.
Treatment of SJIA-related MAS
MAS is treated with anakinra (4–8 mg/kg/day, preferably by intravenous route) monotherapy alone or in combination with glucocorticoids (IV methylprednisolone 20–30 mg/kg/day for 3–5 days followed by oral prednisolone 1–2 mg/kg/day) as and when required on a case-to-case basis with the goal of tapering down to the lowest possible dose in the shortest possible time.
Other agents tried in the treatment of MAS include cyclosporine A, thalidomide, and novel agents such as Emapalumab (anti-IFN-γ), Tadekinig alfa (recombinant IL-18 binding protein), and JAK inhibitors (JAK1/3 inhibitor tofacitinib and JAK1/2 inhibitor Ruxolitinib). 34
Treatment of SJIA – Lung Disease
Management of SJIA-LD includes adequate control of SJIA disease activity, as mentioned earlier, and withdrawal of ongoing biologic therapy if a DRESS-like hypersensitivity reaction is clinically suspected. Other agents tried with demonstrated efficacy in small case reports include JAK inhibitors (Tofacitinib, Baricitinib, and Ruxolitinib), Emapalumab (anti-IFN-γ), intravenous immunoglobulins, plasma exchange, cyclophosphamide, etoposide, mycophenolic acid, and calcineurin inhibitors (Cyclosporine A and tacrolimus). Other lung-directed therapies tried include inhalational glucocorticoids and chronic azithromycin therapy. 34
Treatment of Refractory SJIA – Arthritis
Methotrexate, leflunomide and thalidomide are the conventional immunosuppressive agents used for management of chronic arthritis of SJIA. Anti-IL-6 therapy with Tocilizumab is preferred for initial therapy in the presence of significant arthritis. However, arthritis can be very aggressive and difficult to manage in the absence of inadequate disease control at onset. Other agents tried include Abatacept, Anti-TNF and JAK inhibitors. Although not tried, targeting IL-23-17 axis could be a potential strategy considering the immunopathology of chronic arthritis of SJIA. 34
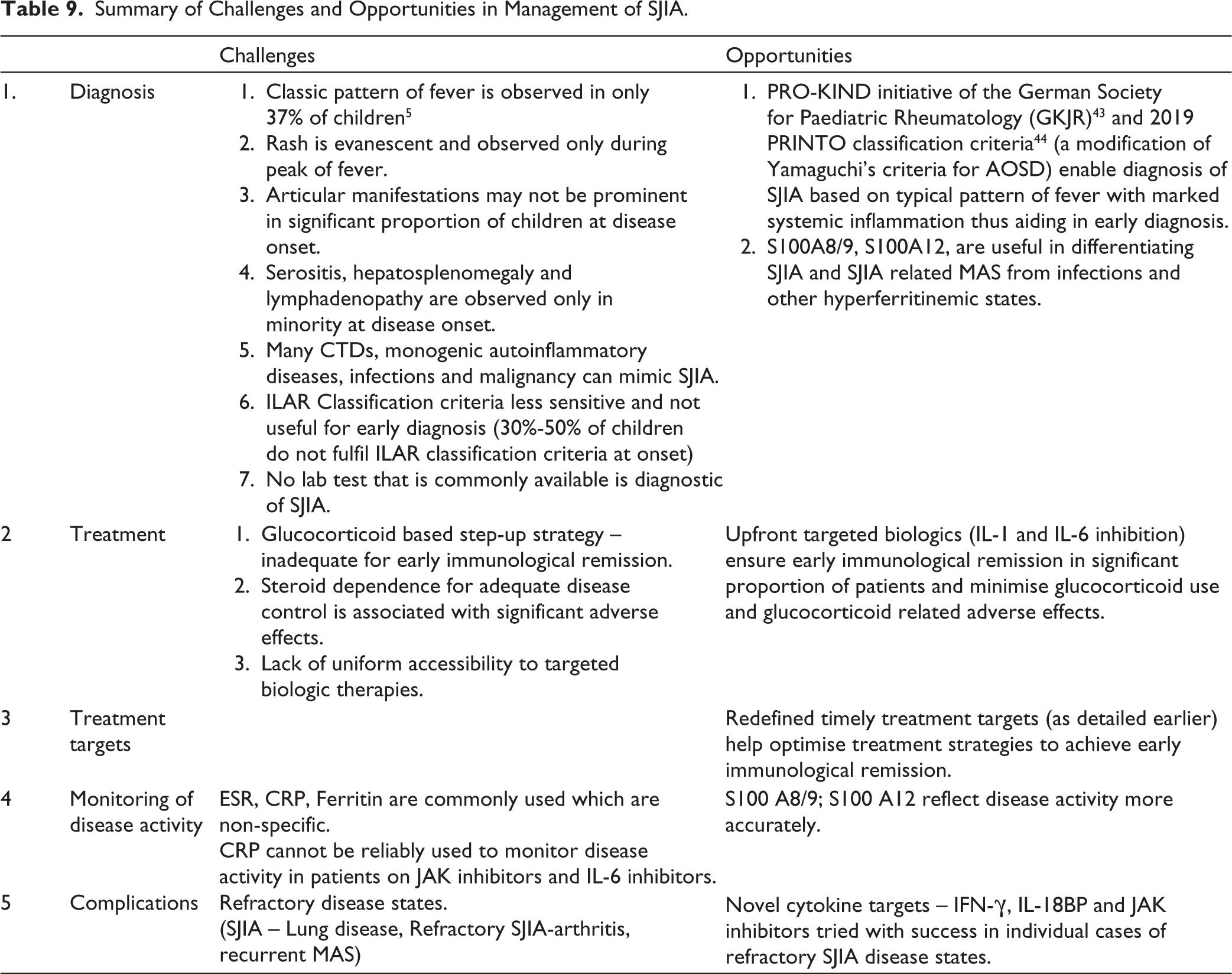
Challenges in the Management of SJIA
The key to effective management of SJIA lies in early diagnosis and prompt control of unchecked innate immune driven inflammation in order to achieve immunological remission and prevent devastating complications. However, heterogeneity in disease presentation, lack of pathognomonic symptoms and signs, lack of uniform access and response to targeted biologic therapy, newly emerging concerns regarding upfront use of biologics, pose a significant challenge in successful treatment of SJIA.
Challenges in Diagnosis
SJIA should be considered in any child presenting with a prolonged, typical quotidian/biquotidian pattern of fever associated with arthralgia/arthritis, typical rash, serositis, lymphadenopathy, or hepatosplenomegaly along with features of marked systemic inflammation.
However, classic pattern of fever is observed in only 37% of children 5 ; rash being evanescent and observed only during peak of fever, is often missed; articular manifestations may not be prominent in significant proportion of children at disease onset; serositis, hepatosplenomegaly and lymphadenopathy are observed only in minority at disease onset. No lab test that is commonly available is diagnostic of SJIA.
The traditionally used ILAR classification criteria, which mandate the presence of arthritis of at least 6 weeks duration with systemic symptoms along with any of the minor clinical features (evanescent erythematous rash, generalised lymphadenopathy, hepatomegaly and/or splenomegaly and serositis) are thus less sensitive and not useful for making an early diagnosis of SJIA. Various international SJIA registries have shown that nearly 30%–50% do not fulfil the ILAR criteria at disease onset, mostly due to a lack of definitive arthritis at symptom onset, and also that any child with arthritis is more likely to be treated before the defined duration of 6 weeks. 5
The different classification criteria used to aid the diagnosis of SJIA are as illustrated in Table 8. Adding to the diagnostic uncertainty, many childhood infections, malignancies, especially acute lymphoblastic leukaemia, connective tissue diseases (CTDs), and monogenic auto-inflammatory syndromes can closely mimic SJIA and it would be prudent to rule them out by appropriate testing before entertaining the diagnosis of SJIA. Hence SJIA still remains a diagnosis of exclusion giving way for diagnostic delay.
Different Classification Criteria For SJIA.

Challenges in the Treatment
Most widely followed treatment strategy, especially in resource-poor settings where accessibility to targeted biologics still remains a challenge, comprises glucocorticoid-based step-up strategy (detailed further in treatment section). This may not be sufficient to achieve early immunological remission and long-term use of glucocorticoids is associated with significant treatment-related adverse effects.
Challenges in Monitoring Disease Activity
Acute-phase reactants such as ESR, CRP, and serum ferritin are commonly used to monitor disease activity which are non-specific. Also, CRP levels may not confidently depict disease activity in a patient treated with tocilizumab or JAK inhibitors and hence are not reliable for monitoring disease activity in such patients.
Newly Emerging Challenges with Use of Upfront Targeted Biologics
With better understanding of disease biology and advent of concept of window of opportunity, a paradigm shift is seen in therapeutic strategy from glucocorticoid-based step-up strategy to upfront targeted biologics. However, with increased use of upfront targeted biologics (IL-1 and IL-6 inhibitors), newer challenges like refractory disease states comprising refractory SJIA arthritis, SJIA – lung disease and recurrent MAS are being noticed, mandating the quest for different therapeutic targets.
Opportunities in Management of SJIA
Early Diagnosis
Considering poor sensitivity of existing ILAR classification criteria in guiding early diagnosis of SJIA, PRO-KIND initiative of the German Society for Paediatric Rheumatology (GKJR) 43 and 2019 PRINTO classification criteria 44 (a modification of Yamaguchi’s criteria for AOSD) were derived which enabled entertaining the diagnosis of SJIA based on a typical pattern of fever and marked systemic inflammation without the need for definitive arthritis. This improved the sensitivity and aided in early diagnosis of SJIA.
Emerging Biomarkers to Aid Diagnosis, Monitor Disease Activity, to Predict Complications and Response to Targeted Therapies
The S100 family of proteins (S100A8/9, S100A12) are calgranulins released by neutrophils. They act as ligands for TLR-4 (pattern recognition receptor), are markedly elevated during the early phase of SJIA, contribute to unchecked inflammasome activation and production of IL-1β and IL-18 and serve as a useful mechanistic biomarker for diagnosis (helps differentiate SJIA from infections) as well as monitoring disease activity after the initiation of therapy. 41 S100 A12 also helps differentiate SJIA-related MAS from other hyperferritinemic syndromes. 46
Elevated IL-18 levels are found during active disease and may also help to predict the evolution of MAS and SJIA-LD. 47
Also, albeit controversial, IL-1RN polymorphisms may serve as biomarkers in predicting response to anti-IL-1 therapy 48 and the HLA-DR B1*15 haplotype as biomarker for predicting hypersensitivity to biologics and the development of SJIA-LD. 49
Change in Therapeutic Strategy
With evolution of concept of ‘window of opportunity’, there is increased emphasis to use upfront targeted biologics (IL-1 or IL-6 inhibitors) targeting early immunological remission, providing an opportunity to prevent crippling articular complications of unchecked SJIA disease activity and long-term glucocorticoid use.
Redefined Treatment Targets
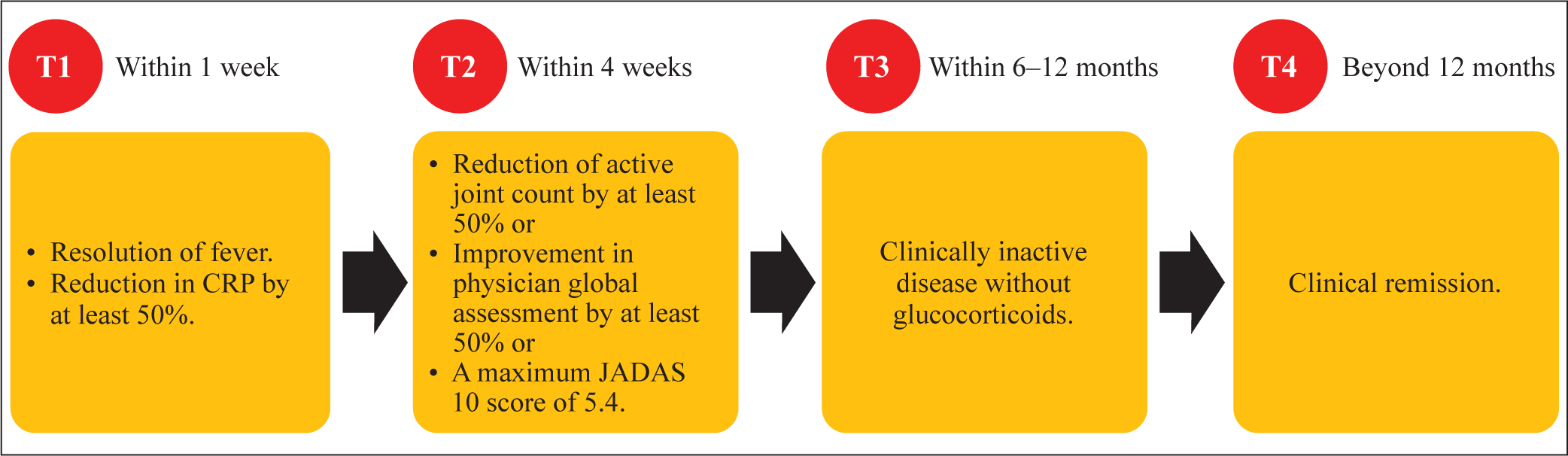
Timely treatment targets are defined by the PRO-KIND initiative, with emphasis on timely treatment escalation, to target early immunological remission (as detailed earlier in Figure 4).
PRO-KIND Initiative Treatment Targets at 1 Week, 1 Month and 1 Year of Treatment.
Novel Cytokine Targets in the Management of Refractory Disease States
JAK inhibitors are being successfully tried in refractory SJIA arthritis, SJIA lung disease and recurrent MAS (JAK1/3 inhibitor tofacitinib and JAK1/2 inhibitor Ruxolitinib). Emapalumab (anti-IFN-γ) and Tadekinig alfa (recombinant IL-18 binding protein), 34 in refractory MAS. Although unexplored, biologics targeting IL-17-23 pathway forms a potential therapeutic target for refractory SJIA-arthritis considering immunobiology of SJIA. The summary of challenges and opportunities in management of SJIA are as illustrated in Table 9.
Summary of Challenges and Opportunities in Management of SJIA.
Conclusion
SJIA remains a distinct type of JIA with predominant innate immune dysregulation and systemic symptoms at disease onset, followed by evolution to adaptive immune dysregulation-driven chronic destructive arthritis. Early diagnosis and adequate, timely control of innate immunity-driven systemic inflammation with upfront anti-IL-1 or IL-6 therapy can lead to immunological remission, minimise steroid use, and prevent the evolution of crippling arthritis and steroid-induced side effects. SJIA can also be associated with potentially fatal complications such as MAS and SJIA-LD and can be refractory to treatment, mandating a continued quest for a deeper understanding of disease biology and trapping novel biomarkers and treatment strategies.
Footnotes
List of Abbreviations
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Funding
The authors received no financial support for the research, authorship and/or publication of this article.