
Abstract
Bradykinin-mediated angioedema can broadly be categorised into hereditary angioedema (HAE) and acquired angioedema (AAE). Both HAE and AAE are grossly under-recognised in the country largely because of lack of awareness. Type 1 and 2 HAE is caused by pathogenic variants in the SERPING1 gene that codes for C1-inhibitor protein. Deficiency of C1-inhibitor protein leads to recurrent swelling episodes involving hands, feet, eyes, lips, tongue and genitalia. These episodes are typically not associated with itching or urticaria. Involvement of the larynx leads to a potentially life-threatening episode of choking and, in the past, the mortality because of laryngeal edema used to be as high as 30%. AAE is usually associated with lymphoreticular malignancies; predominantly B cell lymphomas and systemic lupus erythematosus. Angioedema often precedes the diagnosis of primary illness. The clinical features of both AAE and HAE are similar. However, AAE should be suspected in patients with onset of disease in older age. Management of HAE is broadly categorised into three types: on-demand therapy and short-term and long-term prophylaxis (LTP). Patients with AAE also need management of the underlying disease with immunosuppressants. This review focuses on clinical manifestations, diagnostic evaluation, and clinical mimics of HAE and AAE from the perspective of a rheumatologist. The review also briefly discusses the management principles of HAE and AAE.
Keywords
Introduction
Angioedema is defined as subcutaneous and/or submucosal swelling that is typically localised and episodic. Angioedema is caused by leakage of fluids because of an increase in vascular permeability. It can be broadly classified as mast cell mediator-mediated or bradykinin-mediated.1,2
Bradykinin-mediated angioedema can broadly be categorised into hereditary angioedema (HAE) and acquired angioedema (AAE).1,2 These are uncommon potentially life-threatening disorders. Most cases of HAE (>50%) have onset of symptoms in childhood; however, most often they get diagnosed when they reach adulthood. 3 Family history is reported in approximately 80% of patients with HAE. On the other hand, the onset of symptoms in AAE (in the settings of systemic lupus erythematosus [SLE] or lymphoma) is typically in the fourth to fifth decade of life and without family history.4–6 The cleavage of C1-INH protein by autoantibodies or overconsumption is the cause of symptoms in AAE, rather than deficient or dysfunctional C1-INH, as is seen in HAE. 7 HAE and AAE can be differentiated using C1q protein. In HAE, it remains normal whereas in AAE it is reduced in a large proportion of patients because of increased C1 activation.8,9 These disorders are grossly under-recognised in India, largely because of lack of awareness, and are often misdiagnosed as an allergy.
A large proportion of cases of HAE and AAE get referred to and/or are managed by rheumatologists and immunologists. Hence, they need awareness regarding the clinical manifestations, laboratory diagnosis and therapeutics in these cases.
In this review, we discuss the etiopathogenesis, clinical manifestations and diagnosis of HAE and AAE. We also briefly discuss the management of these disorders.
Epidemiology of HAE in India
The prevalence of HAE worldwide is approximately 1:10,000 to 1:50,000.10,11 The prevalence of HAE in Europe and America is between 1.1 and 1.6 per 50,000. 12 The Asia-Pacific region has a minimal prevalence of HAE at 0.02/100,000 population, with significant heterogeneity in various nations. 13 No national prevalence data for India or other developing countries are available. 13 However, based on the global prevalence of HAE, it has been suggested that India has 27,000 to 135,000 HAE patients. Most of these patients remain undiagnosed.
Etiopathogenesis of HAE
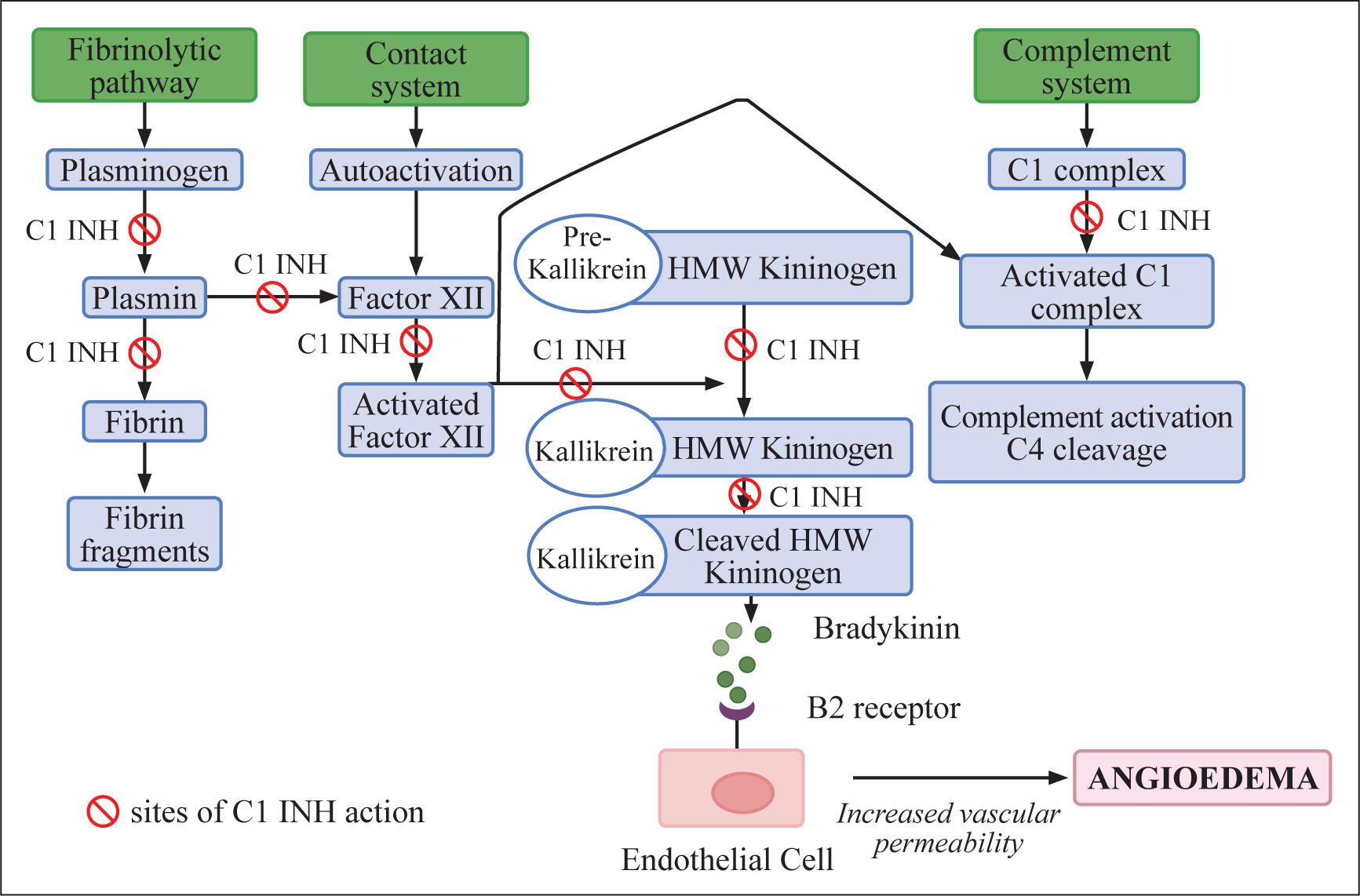
HAE is classified into two types: HAE with C1-inhibitor deficiency (C1-INH-HAE) (Type I caused by reduced levels of C1-INH protein and Type 2, wherein C1INH protein levels are normal but dysfunctional) and HAE with normal C1-inhibitor activity (nl-C1INH-HAE). 2 A mutation in the SERPING1 gene, encoding the C1 inhibitor (C1-INH) protein, accounts for more than 95% of all HAE cases. 14 More than 700 SERPING1 variants have been described. 15 A large proportion (80%) of HAE patients have a suggestive family history as it has an autosomal dominant inheritance and the rest may have a de novo defect in the SERPING1 gene and these patients have no suggestive family history.14,15 C1-INH, serine protease inhibitor (SERPIN), inhibits several proteases, which include complement proteases, contact-system proteases, and fibrinolytic protease. Cleavage of high molecular weight kininogen (HMWK) by activated plasma kallikrein leads to production of bradykinin, a nine-peptide molecule. It is metabolised rapidly by metalloproteases, including angiotensin-converting enzymes (ACE). Factor XII when meets a negatively charged surface gets activated and causes the activation of inactive prekallikrein to kallikrein. C1-INH inhibits both factor XII and plasma kallikrein. Deficiency of C1-INH leads to uncontrolled activation and increased production of bradykinin. Bradykinin acts via B2 receptor and leads to increased permeability of blood vessels leading to edema. 14 (Figure 1) Patients with nl-C1INH-HAE (<5% of all patients with HAE) have been identified to have genetic defects in factor 12 (F12), plasminogen (PLG), myoferlin (MYOF), angiopoietin (ANGPT1), Kininogen 1 (KNG1), and heparan sulfate (HS)-glucosamine 3-O-sulfotransferase 6 (HS3ST6) genes. 2 A small group of patients are categorised as HAE-Unidentified (HAE-U) because despite carrying out extensive investigations, no genetic defect has been identified in these patients. 14 The role of vascular endothelium has also been studied in patients with HAE, and it has been observed that vascular endothelium plays an active role in the pathophysiology of HAE. This has been especially suggested by the identification of defects in angiopoietin (ANGPT1) and myoferlin (MYOF) genes in patients with HAE.14,16 Acquired bradykinin-mediated angioedema is a distinct group wherein the pathophysiology is identical to that seen in HAE, however, no genetic defect has been identified in these patients. Common causes of acquired bradykinin-mediated angioedema include autoimmune diseases and malignancy. Acquired bradykinin-mediated angioedema is more common in adults. 7
Pathogenesis of Hereditary Angioedema: The Deficient or Dysfunctional C1-INH Affects the Fibrinolytic, Contact, and Complement System, Causing an Increase in Bradykinin Production (Binds to B2 Receptors in the Endothelial Cell) and Increased Vascular Permeability.
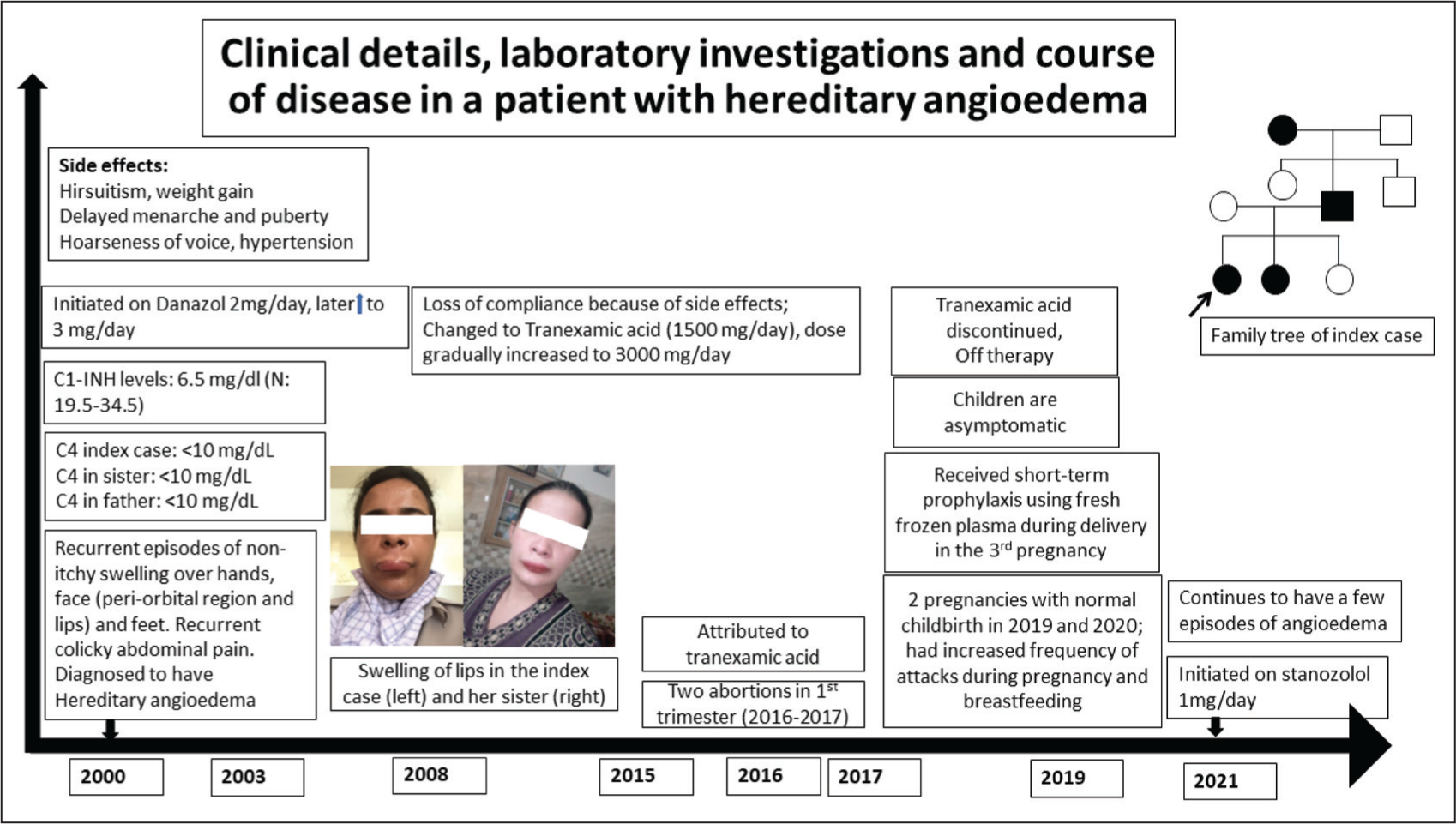
Case Details of the Clinical Course of a Patient with HAE.
A 12-year-old girl presented with non-itchy swelling over the dorsum of her left hand. She had had similar swelling episodes over her face (predominantly over peri-orbital region and lips); hands and feet since the age of 1 year. Family history revealed similar illness in her sister, father, and grandmother. She was diagnosed with hereditary angioedema (HAE) based on low C4, reduced C1-INH levels, and a pathogenic heterozygous mutation (c.51+1 G>A intron 2 splicing defect) in SERPING1 gene. She was initiated on danazol, but as she developed drug-related complications (Figure 1), she was initiated on tranexamic acid (TA). At 27 years of age, she had two first trimester abortions and the cause was attributed to TA use. TA was discontinued. Subsequently, she had two normal pregnancies and childbirths. She experienced increased frequency of episodes of HAE during these pregnancies and during breastfeeding. At the time of caesarean section, fresh frozen plasma was given as short term prophylaxis. She is now on stanozolol and has occasional attacks of angioedema. She was recently given 1000 IU of C1-inhibitor concentrate for an acute episode of laryngeal and facial oedema. She showed prompt improvement in her symptoms. Her children were screened and they were found to be unaffected.
Classification of HAE and Clinical Features
Low functional and antigenic C1-INH results in type 1 HAE, and dysfunctional C1-INH protein leads to type 2 HAE, where C1-INH levels can be normal (or elevated) but have low functional activity. These are the most common types of HAE (>90% of all cases of HAE).2,14
Patients with HAE usually manifest with recurrent episodes of swelling of the hands, feet, eyes, lips, tongue, and genitalia (Box 1) and Figure 2). These episodes are typically not associated with itching or urticaria; they evolve slowly over 12–24 hours and subside over 3–5 days. Gastrointestinal tract involvement can lead to excruciating pain abdomen often associated with abdominal distension, vomiting, diarrhoea, or sometimes constipation. Involvement of the larynx leads to a potentially life-threatening episode of choking and in the past, the mortality because of laryngeal edema used to be as high as 30%. This has considerably reduced with the availability of better first-line treatment options in many countries.2,17 However, mortality due to HAE in India continues to be a major concern. 18
Clinical Details, Laboratory Investigations and Course of Disease in a Patient with Hereditary Angioedema.
Most angioedema episodes in patients with HAE are spontaneous. A proportion of patients may experience triggers for their acute episodes. These include trauma, stress, hormonal changes, and certain foods. Menstruation is a common trigger for females. Estrogen-containing drugs and ACE inhibitors are contraindicated in HAE. 19
Angioedema episodes in HAE is often associated with prodromal symptoms. These include anorexia, alteration in mood, fatigue, malaise, and arthralgia. Erythema marginatum is a specific prodromal symptom that may be seen in 50%–60% of patients in the Western population. 20 In our experience, it has been observed in up to 20% of all patients with HAE.21,22
Acquired Angioedema
Caldwell first described AAE in 1972, and it consists of three components: acquired deficiency of C1-INH, increased activity of the classical complement pathway, and recurrent angioedema episodes.23,24 It is a rare condition, and its prevalence remains speculative due to the absence of epidemiological data. However, it has been hypothesised that the actual prevalence is likely higher, given that the condition is frequently overlooked. 4
Clinically, symptoms of AAE may be difficult to distinguish from HAE. The most important distinction between HAE and AAE lies in the age of symptom onset. Patients with HAE usually become symptomatic within the first 2 decades of life (in >90% of patients), whereas symptoms of AAE manifest usually after the fourth decade. 5 In HAE, angioedema is usually localised to the extremities compared to facial episodes commonly reported in AAE. 25
Pathogenesis of acquired C1-INH defect was initially described in a patient with lymphoma. AAE in lymphoma is due to the depletion of C1-INH or its consumption through over-activation by lymphoma cells.6,26 In 1985, autoantibodies neutralising C1-INH were reported in a subset of otherwise healthy individuals. 4 These anti-C1-INH autoantibodies specifically target epitopes surrounding the reactive centre of C1-INH. Their binding to these epitopes results in the functional inactivation and/or increased catabolism of the protein. These autoantibodies are reported in patients with SLE and lymphoproliferative disorders. 27
AAE typically manifests in individuals at an advanced age. In a study from the United States, AAE was shown to be associated with lymphoreticular malignancies, predominantly of B cell neoplasms, particularly non-Hodgkin’s lymphoma (NHL), followed by splenic marginal zone lymphoma, acute leukaemia, and Hodgkin’s lymphoma. In 6 of the 7 patients, AAE was diagnosed prior to the diagnosis of malignancy. 28
A minority of patients with AAE exhibit monoclonal gammopathy of undetermined significance (MGUS). Bozek et al. conducted a study examining various neoplastic disorders, such as papillary thyroid carcinoma and breast carcinoma. The development of AAE in lymphoproliferative disorders involves either C1-INH consumption or the formation of idiotype or anti-idiotype autoantibodies, the latter being more prevalent.29,30
SLE is another common cause of AAE, although less common than malignancies. Key features pointing towards AAE in these patients include later onset, absence of familial history, and low C1q levels. 27 AAE associated with SLE may be due to low levels of complements C3 and C4 due to increased consumption rather than because of anti-C1-INH autoantibodies. The incidence of angioedema in patients with SLE varies based on ethnicity and gender, with a higher prevalence in African Americans, followed by Asians, and more commonly observed in females. Out of 19 patients of SLE with angioedema reported in a review, SLE was diagnosed in 8 patients at the time of development of angioedema while others had already been diagnosed to have SLE at the time of onset of angioedema.27,31
A systematic review by Shi et al., involving 154 patients with AAE, reported a median age of symptom onset at 64 years, with face being the primary site of attacks in 85% of patients. 7 Low levels of C4, C1-INH antigen, and C1-INH function were observed in almost all patients, along with low C1q levels in >65% of cases.4,7 Although AAE is rarer than HAE, the diagnostic delay is surprisingly shorter (average 7.5 months). It was observed that in one-third of patients, the diagnosis of angioedema preceded the diagnosis of primary illness and others were diagnosed either at the time of diagnosis of primary illness or later. There was no difference in the age, area involved or potential triggers of angioedema in patients based on the underlying etiology. However, a significant difference was observed based on sex, with more female predilection seen in patients with lymphoproliferative disorders (P = 0.003). The diagnostic delay was longer who had solid tumours or autoimmune disorders (P = 0.049) and significantly low C1-INH autoantibodies were seen in lymphoproliferative disorders (P = 0.003).
Another study reported that in 90% of patients the symptoms of angioedema were present before the diagnosis of their underlying disease (Lymphoma, SLE).27,30
Clinical Mimics of HAE and AAE That a Rheumatologist Should Be Aware of?
It’s important to remember that not every swelling is angioedema. Rheumatologists must understand the common differential diagnoses of HAE and AAE. The most common diseases that resemble angioedema are urticarial vasculitis, drug rash, eosinophilia and systemic symptoms (DRESS) syndrome, orofacial granulomatosis, urticarial vasculitis, and infections. These conditions do not respond to the usual forms of treatment used for angioedema.32–34
Urticarial Dermatitis (UD)
UD is a severe pruritic skin eruption that can appear in the form of papules and plaques like urticaria. UD typically persists for more than 24 hours, unlike urticaria, which resolves quickly. UD can last for days to weeks. The trunk and extremities are common distribution points, and palms and soles are usually spared. Chronic scratching can lead to lichenification. When distinguishing features of other dermatoses are absent, a skin biopsy can be useful. 35
Urticarial Vasculitis (UV)
UV is characterised by angioedema, urticaria, and necrotising vasculitis. It is of 2 types: hypocomplementemic and normal complement levels. Urticaria and chronically acquired hypocomplementemia are symptoms of hypocomplementemic UV (variably low C3 and low C4). Approximately 50% of patients develop angioedema, primarily in the lips, tongue, periorbital tissue, and hands, and may be the presenting symptom. These skin lesions are usually painful and heal with post-inflammatory hyperpigmentation. 36 At times, patients with IgA vasculitis (Henoch-Schonlein purpura) may present with edema of the hands and feet associated with abdominal pain with a very faint rash. The swelling may mimic angioedema and may lead to misdiagnosis.
Infections
Infections of the periorbital area, gingiva, and tongue mimic angioedema and persists until infections are managed. Methicillin-resistant Staphylococcus aureus-related facial or lip infections resembling angioedema have been reported. Tongue abscess causes enlargement of tongue and may present like angioedema. Periorbital edema due to trichinosis and tropical filariasis are frequently misdiagnosed as angioedema.37,38
Dress
This is a life-threatening condition that occurs approximately 2–6 weeks after intake of the offending drug. This cutaneous condition has a mortality rate of 10%. Maculopapular rash with diffuse skin edema, and facial edema may be the presenting features and can be mislabelled as angioedema. History of drug intake, presence of organomegaly and lymphadenopathy, eosinophilia, and raised IgE levels help clinch the diagnosis. 39
Orofacial Granulomatosis
Granulomatous lesion of the maxillofacial, oral and anogenital region leads to swelling of the local area mimicking angioedema. Persistent and chronic nature of the edema and characteristic histopathological examination helps in differentiating it from angioedema. Melkersson-Rosenthal syndrome is characterised by persistent painless swelling of the lips and face, frequently associated with fissured tongue and facial palsy. Isolated labial involvement may also mimic angioedema.37,40
Hypothyroidism
Patients with hypothyroidism may present with persistent periorbital swelling.
Diagnosis of HAE and AAE
Diagnosis of HAE and AAE relies on a good clinical history and a strong clinical suspicion. The suspicion of HAE should be made in a patient who has a history of recurrent swelling of skin (hands, feet, face, and genitals), gastrointestinal episodes (abdominal pain), and laryngeal swelling without any itching or urticaria. 25
Clinical suspicion of HAE would be strengthened by the presence of one or more of the following features:
Suggestive family history Early age of onset of clinical features Unresponsiveness to antihistamines, adrenaline and glucocorticoids Presence of trigger and prodrome
The international guidelines for the diagnosis and management of HAE propose testing for levels of C4 and C1-INH along with C1-INH function in every patient with suspicion of HAE. Patients with type 1 HAE have low C4, low C1-INH levels, and low C1-INH function while patients with type 2 HAE have low C4, normal (or high) C1-INH levels and low C1-INH function. 2 However, availability and access to all three tests for screening HAE patients in India is a challenge. Hence, the recently published consensus statement from India suggests using C4 levels to screen patients with HAE. Assessment of C4 alone as a screening test for HAE has approximately 80% sensitivity with normal C4 levels in up to 20% of patients with type 1 and 2 HAE.25,41 The sensitivity of the C4 test, however, can be increased if the test is done during an acute episode. A proposed algorithm for the diagnosis of HAE is given in Figure 3.
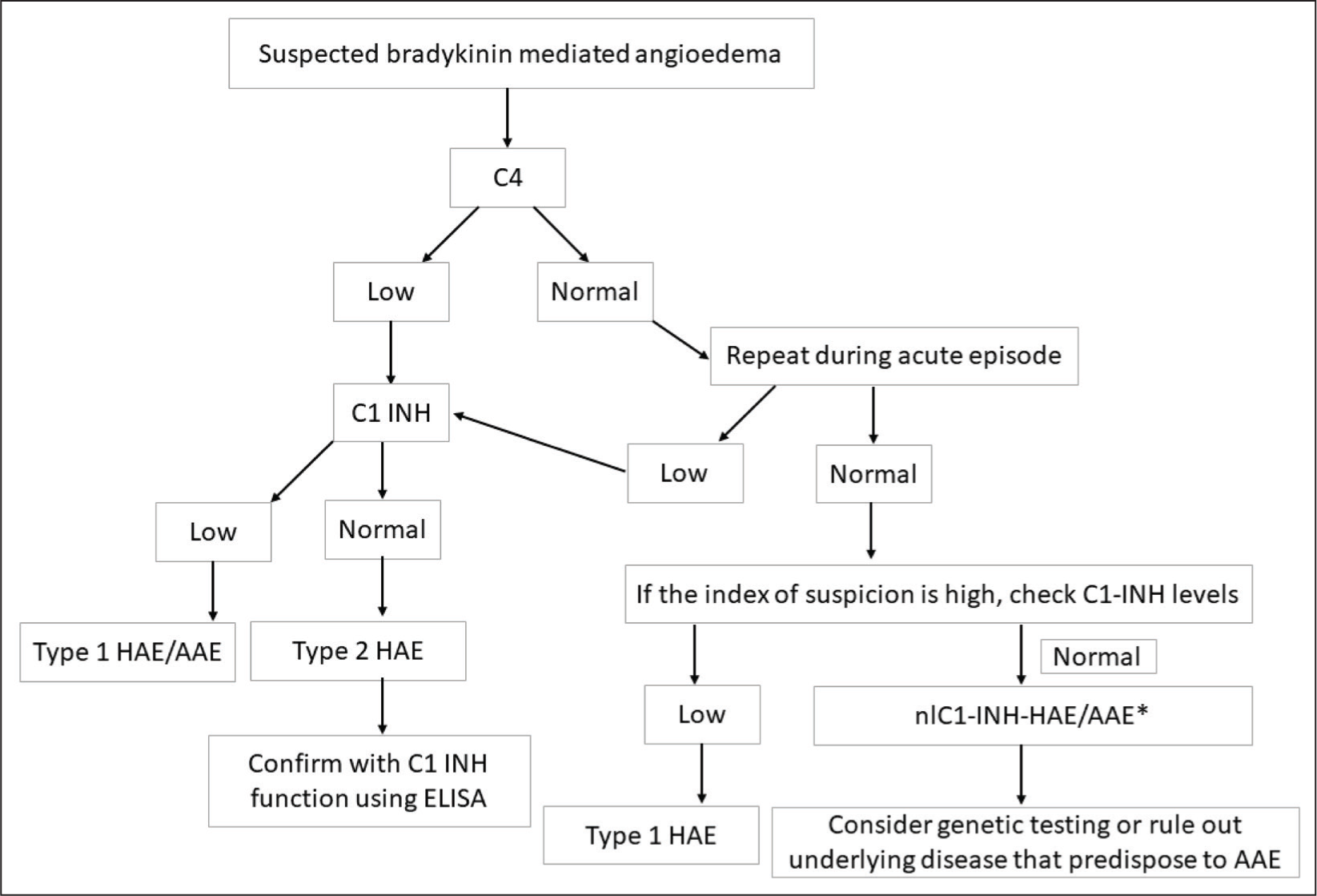
C4 and C1-INH levels are usually assessed by nephelometry while functional assay for C1-INH is carried out by enzyme-linked immunosorbent assay (ELISA). 25
Patients with ACE inhibitor-induced angioedema and idiopathic acquired AAE have normal levels of C4, C1 INH, and normal C1-INH function.
AAE should be suspected in all patients who have symptoms consistent with bradykinin-mediated angioedema and have onset of disease after fourth decade of life. Approximately 70% of AAE patients have low C1q levels (in addition to having reduced C4, reduced C1-INH levels and decreased C1-INH function) which is one of the main laboratory findings that differentiates it from HAE as C1q levels are normal in HAE. 38
All patients with AAE need to be evaluated for lymphoproliferative and autoimmune diseases. A complete blood count including differential white blood cell counts, antinuclear antibodies, chest X-ray, along with abdominal ultrasound should be done in all cases at diagnosis of AAE and should be repeated annually as patients may develop symptoms of autoimmune diseases or lymphoproliferative disease a few years after the diagnosis of AAE.7,30
Genetic test is not mandatory to diagnose a patient with HAE. However, genetic testing is required for antenatal diagnosis, for diagnosing HAE in young infants (where complement studies may not be reliable), and for diagnosing normal C1-INH HAE (previously known as type 3 HAE). Next-generation sequencing is the preferred technique used for establishing the genetic diagnosis.22,42
Management of HAE
The primary goal of managing HAE is to prevent mortality related to the disease and improve the quality of life. The availability of better first-line therapeutics in developed countries has significantly reduced mortality associated with HAE. However, when first-line medications are not readily available, mortality remains a concern. 18 Plasma-derived C1-INH (pd C1-INH) concentrate is now available in India and is increasingly used for management of acute episodes.
This review is not intended to discuss the details of the management of HAE and AAE. However, basic principles of management will be discussed.
Management of HAE is based on three key principles (Table 1):
Drugs Used in the Management of HAE.

Treatment of Acute Attacks (i.e., On-demand Therapy)
On-demand therapy focuses on addressing acute attacks to alleviate symptoms and minimise their impact. Ideally, all angioedema attacks need on-demand therapy. The drugs that are used for on-demand therapy are:
pd C1-INH concentrate: Preferred choice for managing acute attacks in HAE in many developed countries. 2
Ecallantide (plasma kallikrein blocker) and Icatibant (competitive antagonist of B2 receptor) are indicated for on-demand treatment. 2 However, these are not available in India currently.
Fresh frozen plasma (FFP) (10–20 mL/kg, containing 1 unit of C1-INH per ml) can be used for the management of acute attacks of HAE. Due to potential side effects including infusion reactions, anaphylaxis, risks of transmission of infections along and volume overload, it is recommended to be used in acute life-threatening situations such as laryngeal edema. 2
Short-term Prophylaxis
Short-term prophylaxis is needed in situations where there is a predictable risk of developing life-threatening laryngeal edema. High-risk situations involving laryngeal manipulation, such as those requiring general anaesthesia and laryngeal intubation, warrant short-term prophylaxis. pd C1-INH concentrate is the best possible option for STP in India and should be administered soon before the intervention (dose 10-20 units per Kg). The dose may be repeated, if required.2,25,43
FFP and attenuated androgens in combination may also be used for STP. For patients who are taking attenuated androgens as long-term prophylaxis, doubling the dose 2 days before the anticipated procedure and continuing for 5 days post-procedure is recommended. In cases where patients are not using attenuated androgens, it should be initiated two days before the procedure (Dose: stanozolol 2 mg/day) and continued for five days post-procedure. FFP administration (@ 10 mL/kg twice daily) is recommended 1–2 days before the procedure and a single dose on the day of the procedure. FFP administration post-procedure may be considered on a case-to-case basis, especially for patients undergoing laryngeal manipulation.41,43
Long-term Prophylaxis
LTP includes administration of drugs on a continuous basis to prevent development of attacks or to reduce their severity, especially for patients with frequent and debilitating episodes.
Tranexamic Acid
Tranexamic acid, an antifibrinolytic agent, inhibits plasmin production, reducing bradykinin production. While less effective than attenuated androgens, the better safety profile of tranexamic acid makes it a preferred choice for LTP especially in children, adolescents, and pregnant women (if required). A 30–50 mg/kg/day dose in 2–3 divided doses (maximum dose 3 g/day) is recommended. 2
Attenuated Androgens
These drugs may increase C1-INH production and are commonly used for LTP. Stanozolol (dose 0.5 mg every other day to 4 mg/day) and danazol (100 mg every other day to 600 mg/day) are two common attenuated androgens used for LTP. Dose-limiting side effects include various complications and a close watch on growth and blood pressure. Monitoring of liver enzymes, and liver ultrasound also becomes necessary during chronic use of the drug.2,13
Newer Agents
Lanadelumab (300 mg subcutaneous 2–4 weekly) is a fully humanised monoclonal antibody targeting plasma kallikrein and has been approved for LTP in adolescents and adults.2,44
Berotralstat (BCX7353) is another promising LTP. It is a highly selective inhibitor of plasma kallikrein. 2 In phase 3 randomised controlled trial, orally administered berotralstat 150 mg once daily decreased the frequency of HAE attacks significantly and exhibited a favourable safety profile. 20
An in vivo gene-editing therapeutic called NTLA-2002 is based on CRISPR (clustered regularly interspaced short palindromic repeats) and CRISPR-associated protein 9. With just one dosage, NTLA-2002 aims to permanently suppress angioedema episodes by targeting the kallikrein B1 (KLKB1) gene. No serious side effects were reported and total plasma kallikrein was effectively suppressed. 45
Other General Measures
Identification of potential triggers and counselling to avoid them is essential for the management of patients with HAE. ACE inhibitors and estrogen-containing pills must be avoided in all patients.
Management of AAE
A similar approach to management has also been recommended for patients with AAE. Till date, there is no known cure for HAE, however, patients with AAE may respond to the management of the underlying disease. It is also to be noted that treatment of the underlying disease may not always improve AAE symptoms.7,27,45
Off-label treatment of AAE is based on treatment principles for HAE, and several studies have reported good results and positive outcomes from acute treatment as well as long-term prophylaxis. Rituximab has shown promising results in patients with AAE including those without lymphoproliferative diseases or MGUS. 30 However, in the absence of evidence of lymphoproliferative disease or MGUS and considering the safety profile of medications used to treat angioedema, the decision to initiate an immunosuppressant should be considered carefully.7,28,30,38
While pd-C1-INH is beneficial in most patients with AAE, some may develop resistance to it or require higher dosages, especially those with anti-C1-INH antibodies. In patients with anti-C1-INH autoantibodies, the dose required to effectively treat angioedema episodes may be more than double the dose required in patients without autoantibodies. The response to C1-INH therapy varies from patient to patient and across attacks. This could be due to differences in the levels of anti-C1-INH autoantibodies between attacks. Repeated, regular and long-term testing of anti-C1-INH autoantibody levels should be done in AAE patients to titrate the dose.
While long-term prophylactic treatment is commonly used in HAE, its use in AAE has yielded mixed results. Androgen derivatives that are very effective in HAE, may not be as successful in AAE, possibly due to the rapid catabolism of C1-INH. On the other hand, antifibrinolytic agents used for prophylaxis are more effective in AAE than HAE. This could be attributed to their anti-plasmin activity, which is crucial for angioedema symptoms in AAE.7,28,29,47,48
A systematic review by Shi et al. on the outcome of patients with AAE reported mortality in 9 patients (7.4%) and remission in 74 patients (64.3%) with a 10-year survival as high as 79.2%. Factors that play an important role in causing disease remission were found to be age, male sex, presence of monoclonal gammopathy and requirement of specific on-demand treatment.
Conclusion
To conclude, HAE and AAE are grossly under-recognised in India and many patients may present for the first time to a rheumatologist. It may be difficult to clinically differentiate HAE and AAE. Patients with AAE typically have onset of symptoms during 4th to fifth decade of life while most HAE patients have onset in the first two decades of life. Lymphoproliferative disorders and autoimmune diseases (such as SLE) are two common underlying conditions that predispose to develop AAE. C4 is a simple screening test for HAE and AAE. pd C1-INH concentrate is now available in India and is a useful drug for on-demand treatment and STP in HAE and AAE.
Footnotes
Authors Contributions
All authors contributed to the study’s conception and design. The first draft of the article was written by Suprit Basu and Reva Tyagi. Ankur Jindal and Sanghamitra Machhua commented and corrected on previous versions of the article. All authors read and approved the final article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Funding
The authors received no financial support for the research, authorship and/or publication of this article.