
Abstract
Background
Lung cancer is a highly lethal malignancy that affects millions of people worldwide. Treatment remains challenging, particularly due to tumor invasion and metastasis. While previous studies have explored various therapeutic approaches, the multifaceted mechanisms of triptolide in lung cancer treatment have not been comprehensively reviewed.
Objectives
This study aims to provide a systematic and in-depth analysis of recent research on triptolide and its derivatives, focusing on their anti-lung cancer mechanisms.
Methodology
A thorough review of the literature was conducted to examine the effects of triptolide on lung cancer cells, including its roles in inducing apoptosis, enhancing immune response, inhibiting invasion and migration, combating oxidative stress, reducing inflammation, and reversing therapy resistance.
Results
Triptolide exerts significant anti-tumor effects through multiple pathways, not only via established mechanisms such as apoptosis induction and metastasis suppression, but also through novel processes like cuproptosis (copper-dependent cell death). It also modulates inflammatory factors and immune regulation within the tumor microenvironment.
Conclusion
This review offers new perspectives on triptolide’s anti-cancer mechanisms and provides a valuable scientific foundation for the development of novel therapeutic strategies and drugs for lung cancer treatment.
Introduction
The most recent global cancer statistics show that lung cancer is the deadliest type of cancer, killing around 18% of all cancer fatalities. Every year, around 18 million people find out they have lung cancer for the first time, and roughly 90% of them die (Sung et al., 2021). In China, lung cancer is more common than other types of cancer and is now the leading cause of mortality, which is a severe threat to people’s health (Hirsch et al., 2017). The spread and invasion of lung cancer cells is a major cause of death in patients. Around 90% of cancer fatalities are caused by cancer cells spreading and invading other cells. In recent years, there has been some progress in the clinical treatment of lung cancer. However, stopping lung cancer cells from invading and spreading is still a difficult problem that involves many processes, such as resistance to apoptosis, degradation of the extracellular matrix, epithelial–mesenchymal transition, angiogenesis, immune escape, cancer cell migration and invasion, and abnormal expression of the tumor microenvironment. So, it is really important to identify innovative ways to treat lung cancer and stop it from spreading and invading other parts of the body.
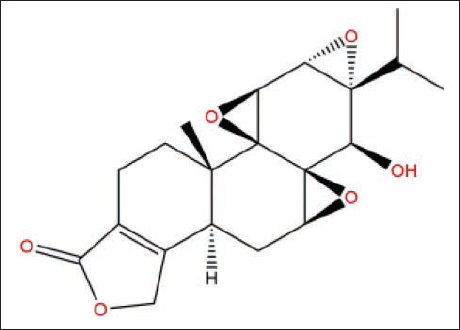
Tripterygium wilfordii Hook.f. is a traditional Chinese medicine made from the xylem of the T. wilfordii Hook.f. plant (Celastraceae). People in China have used this therapeutic substance for a long time. The “Southern Yunnan Herb,” put together by Lan Mao in the Ming Dynasty, was the first place it was seen. The text says that it is hot, mild, and poisonous, and that it can influence more than one meridian system. The “Hunan Medicinal Records” also say that T. wilfordii can kill insects, reduce inflammation, and get rid of toxins (Miao et al., 2019). Triptolide (TP) is an epoxidized diterpene lactone that comes from the plant T. wilfordii. As illustrated in Figure 1, a recent scientific study has established that it has many pharmacological properties, including anti-inflammatory (Gao et al., 2021, 2022), anti-fertility (Liu, 2011), and immunosuppression (Bai et al., 2016; Yuan et al., 2019). Researchers are very interested in triptolide since recent studies have shown that it has a strong inhibitory effect on more than 60 tumor cell lines, including those for cervical, breast, and lung cancer.
Molecular Structure of Triptolide.
TP-related Classical Signaling Pathways
P53-mediated Pathway
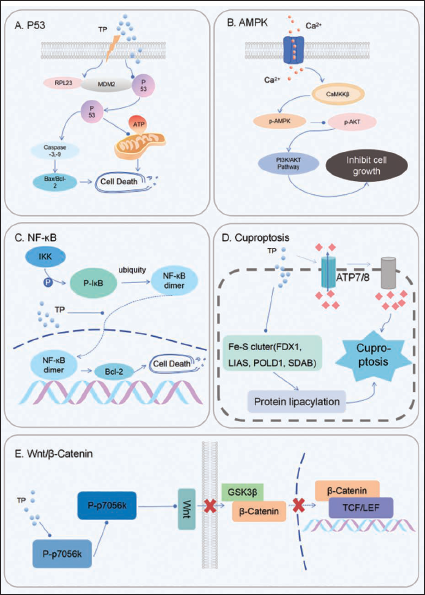
Since it was found in 1979, the p53 gene has been known to be a significant tumor suppressor (Sak et al., 2003). The p53 gene changes the ratio of Bax to Bcl-2 by positively regulating the pro-apoptotic gene Bax and negatively regulating the anti-apoptotic gene Bcl-2. This allows for precise control of the apoptosis process. Researchers (Wang et al., 2020) observed that TP can help ribosomal protein L23 (RPL23) stick to the mouse double minute gene (MDM2). RPL23 and p53 both bind to MDM2, and they compete with each other for that binding. When TP made RPL23 connect to MDM2 more tightly, the p53 was attached to MDM2 less tightly, which made the amount of unbound p53 go up. This rise in free p53 turns on caspase-3 and caspase-9, which starts a chain reaction that raises the Bax/Bcl-2 ratio and causes A549 cells to die. Researcher (Kumar et al., 2016) revealed that TP controls the level of deacetylase 3 (SIRT3) by using p53. This impacts the mitochondrial pathway, which controls cell respiration and ATP production, and helps fight lung cancer. TP can boost the transcriptional activity of heat shock protein 70 (HSP70). It can also break down the unfolded mutp53 protein and the folded mutp53 protein, which stops non-small cell lung cancer (NSCLC) from spreading. So, the technique of getting rid of mutp53 protein could be very helpful for treating NSCLC that has a TP53 mutation (Figure 2A).
Classical Signaling Pathways That are Connected to Triptolide (TP). (A) TP Stops the P53 Pathway, Which Allows It to Control the Apoptotic Process Very Precisely. (B) TP Helps Lung Cancer Cells Die by Controlling the PI3K/Akt Pathway and the Levels of Proteins That are Involved in Apoptosis. (C) TP Stops NF-κB from Working and Kills Cells. (D) TP Raises the Amount of Copper Ions Inside Cells, Which Causes Acylated Proteins to Clump Together in an Unusual Way Inside Mitochondria and Disrupts Iron–Sulfur Cluster Proteins That are Important for Mitochondrial Respiration. This Process Starts a Stress Response in Proteins That Makes Cells Die. (E) TP Stops the p70S6K/GSK3/β-catenin Signaling Pathway, Which Causes Lung Cancer Cells to Die.
Inhibition of the AMPK/PI3K/AKT Signaling Pathway
AMPK, or AMP-activated protein kinase, is a heterotrimeric protein made up of three parts: α, β, and γ. The AMPK/PI3K/AKT signaling pathway is very important for controlling cell growth, division, death, the inflammatory response, metastasis, and keeping the cancerous biological traits of tumor cells. Because of this, blocking this route has become a possible target for cancer treatment (Lin et al., 2022). According to Xie et al. (2016), TP can increase the levels of p-P38, p-ERK, and Bax, while lowering the levels of p-JNK, p-Akt, and Bcl-2. The activation of p38 and the low expression of JNK are two of the things that help make tumor suppressor genes. The higher phosphorylation level of ERK can lead to apoptosis, and the abnormal activation of Akt is closely linked to drug resistance and tumor prognosis. This means that TP promotes the apoptosis of A549 cells by controlling the PI3K/Akt pathway and the levels of proteins that are linked to apoptosis. Dai et al. (2021) found that as the concentration of TP rose, the phosphorylation level of Akt fell, the phosphorylation level of AMPK rose, and the intracellular Ca²+ concentration rose a lot, which turned on Ca²+-dependent kinase (CaMKKβ). This suggests that the CaMKKβ/AMPK signaling pathway may need to be turned on for TP to cause apoptosis. The PI3K/Akt pathway can stop bronchial epithelial cells from autophagy and make lung damage worse, according to Zhu et al. So, stopping this signaling pathway may be very crucial for treating lung cancer and reversing medication resistance (Figure 2B).
Inhibition of NF-κB Transcriptional Activity
TP has been shown to work well as a blocker of NF-κB activation (Liu et al., 2000). In the study of pancreatic cancer xenografts, low oxygen levels are a sign of invasive development and spontaneous metastasis (Chang et al., 2011; Geismann & Arlt, 2020). TP therapy can lower the activity of NF-κB binding that is caused by low oxygen levels. This stops the expression of its subunits, especially the c-Rel and Rel-A proteins. TP binds to p38α and extracellular regulated protein kinase 1/2 (ERK1/2) and makes them work (Park, 2014; Zheng et al., 2017). It causes p53 to be phosphorylated and stabilized. p53 then competes with IκBα, which stops NF-κB, to attach to IKKβ. This process stops IκBα from being phosphorylated and broken down, which stops NF-κB from moving into the nucleus. Using TP breaks the H19/MiR-204-5p/NF-κB/FLIP axis, which causes FLIP to break down without the help of proteasomes and tumor necrosis factor-α (TNF-α) to kill tumor cells (Liu et al., 2022; Yuan et al., 2022). When this axis was disrupted, TP’s anti-cancer action got a lot stronger. When cells get signals of stress or inflammation, NF-κB turns on. NF-κB is often always active in several types of malignancies, according to research (Li, Qin et al., 2024). Jiang et al. (2016) discovered that TP might make A549/TaxR cells less resistant to drugs by blocking the NF-κB signaling pathway and the expression of drug resistance genes that it controls (Figure 2C).
Inducing Copper Death of Lung Cancer Cells
Copper death is a way for cells to die when there are too many copper ions inside them. It makes acylated proteins clump together in mitochondria in an unusual way, and it messes up iron–sulfur cluster proteins that are important for mitochondrial respiration. This makes proteins poisonous and kills cells (Tsvetkov et al., 2022; Wang et al., 2022). Copper death mostly affects mitochondria. The JC-1 fluorescent probe showed that TP changed the mitochondrial membrane potential in A549 and H460 cells. Western blot examination showed that treatment with TP lowered the levels of iron–sulfur cluster proteins (FDX1, LIAS, POLD1, SDHB) and the DLAT monomer, but raised the levels of DLAT oligomers. FDX1 and LIAS are important for adding acyl groups to proteins in iron–sulfur clusters. DLAT is a protein in the pyruvate dehydrogenase complex that has an acyl group attached to it. Too much copper causes DLAT to oligomerize, which is poisonous and kills cells (Solmonson et al., 2018; Tang et al., 2022; Tian et al., 2013). These results imply that TP may stop lung cancer cells from growing and moving by causing copper death (Figure 2D).
Other Apoptotic Pathways
According to Tian et al. (2021), as the concentration of TP rose, the levels of β-catenin, p-p70S6K, p-GSK-3α, p-GSK-3β, Jagged1, and c-Myc fell. The Wnt/β-catenin signaling system depends on β-catenin, and p70S6K and GSK-3 stop β-catenin from working. When GSK-3β is turned on, it can help break down β-catenin protein, which stops it from doing its job of transcribing. So, by stopping p70S6K from working, you can stop the Wnt/β-catenin pathway, which will turn on GSK-3 and speed up the breakdown of β-catenin. This study showed that TP can kill A549/TaxR cells by blocking the p70S6K/GSK3/β-catenin signaling pathway (Figure 2E).
The Intervention Effect of TP on Immune Escape of Lung Cancer Cells
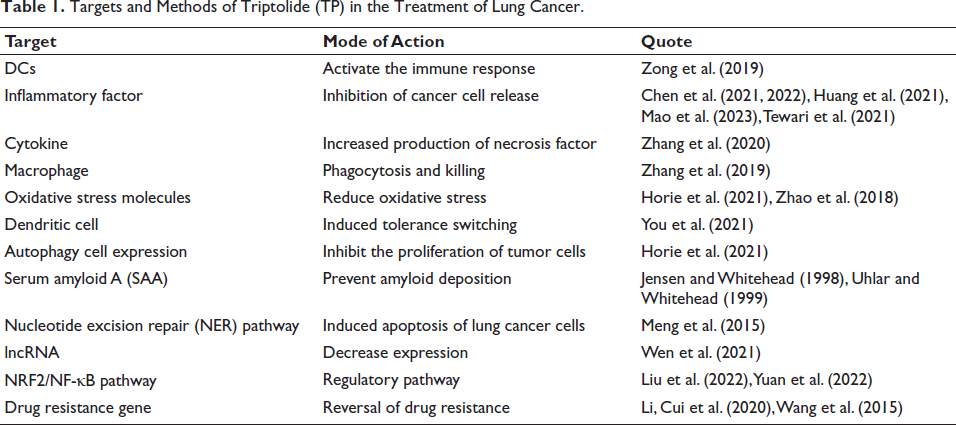
The effect of TP on lung cancer cells’ ability to bypass the immune system TP can help cure lung cancer by speeding up the maturation and activation of dendritic cells (DCs), which are important immune system cells that present antigens. This makes it easier for T cells to respond to tumors, which activates the immune system. TP also boosts the immune response by controlling the release of cytokines, like boosting the production of TNF-α and interferon-γ (IFN-γ). These cytokines can help immune cells become active and multiply, and they can also make tumor cells better at destroying. Also, TP may stop tumor cells from stopping the immune system and bring back T cells’ ability to fight tumors by changing how immune checkpoint molecules like programmed death protein 1 (PD-1) and its ligand (PD-L1) are expressed (Zong et al., 2019). This rule about immunological checkpoints helps the immune system find and get rid of cancers better by stopping tumor cells from escaping the immune system. TP may stop the activation of IRF3, which lowers the amount of type I IFN that is overexpressed (Zhang et al., 2015). This could help keep the immune system from going overboard and causing too much inflammation and damage, while still keeping a reasonable anti-tumor response. TP can also control the polarization of macrophages, push them toward the anti-tumor M1 type, and make them better at eating and destroying tumor cells (Li et al., 2025).
TP Antioxidant
TP causes oxidative stress by raising the amount of reactive oxygen species (ROS) in lung cancer cells, which throws off the balance of redox inside the cells. This oxidative stress can hurt macromolecules inside cells, such as proteins, lipids, and DNA. This can turn on stress responses and signaling pathways inside cells, like the mitogen-activated protein kinases (MAPKs) and phosphatidylinositol 3-kinase/protein kinase B (PI3K/Akt) pathways.
Nrf2 is a critical player in activating intracellular antioxidant responses. It is also important for cellular defense mechanisms since it boosts exogenous metabolism and the production of cytoprotective enzymes (Zhao et al., 2018). Keap1 is part of the CUL3-dependent E3 ubiquitin ligase complex and controls how Nrf2 is activated and how stable it is. Nrf2 and Keap1 come together to form a trimer, and Nrf2 speeds up the breakdown of Keap1 by boosting the ubiquitination of the Nrf2 Neh2 domain (Horie et al., 2021). When there is stress, Keap1 breaks down and activates Nrf2. This protein then goes into the nucleus and starts the production of antioxidant enzymes like HO-1 and NQO1 to keep the balance of everything inside the cell. HO-1 is an enzyme that protects cells by getting rid of toxic heme and making bilirubin, iron, and carbon monoxide. It protects cells by lowering oxidative stress, controlling inflammation, and encouraging angiogenesis. NQO1 is a flavinase that is controlled by Nrf2 and has an ARE sequence. It is upregulated in the antioxidant response along with other antioxidant enzymes like HO-1. TP treatment of lung cancer cells can greatly boost the production of Nrf2 and its downstream proteases NQO1 and HO-1. This shows that TP may have an antioxidant impact (You et al., 2021).
TP-induced Cell Death
The G2 phase is very important for cells to stay alive. If there are less G2 phase cells, it can make damaged tumor cells go through mitosis and perish (Zhang et al., 2020). TP changes the distribution of cells in the cell cycle in A549/Tax9 cells by increasing the number of S-phase cells and decreasing the number of G2/M-phase cells. This stops the S-to-G2 transition. TP also controls the amount of calcium (Ca²+) inside cells by turning on the Ca²+/CaMKKβ/AMPK pathway, which increases AMPK phosphorylation and decreases AKT phosphorylation at the S473 and T308 sites (Wang et al., 2020). This process eventually leads to cell death. When you silence Drp1 (a protein that helps divide mitochondria) in HCT-116 colorectal cancer cells, it stops apoptosis after TP therapy.
Bcl-2 and Bax are two important proteins that control mitochondrial apoptosis. Bcl-2 stops apoptosis, while Bax starts it. Caspases are closely linked to the death of eukaryotic cells. TP stops A549 cells from growing by making them more likely to die, lowering the amount of Bcl-2 mRNA, and raising the amount of Bax, caspase-3, caspase-8, and caspase-9 mRNA (Zhao et al., 2018). This shows that TP causes tumor cells to die by changing the levels of proteins that are involved in apoptosis.
Autophagy is an important way for cells to break down and recycle things, and it is linked to the growth of tumors. Beclin1, LC3B, and P62 are important proteins that help with autophagy, and their levels show how active autophagy is (Zhang et al., 2019). TP stops the growth of A549 cells by raising autophagy and the levels of LC3B, Beclin1, and P62. This shows that it can stop the growth of tumor cells by controlling proteins that are involved in autophagy.
TP Reduces Inflammation
Inflammation plays a big role in how tumors grow, as cells and chemicals work together in complicated ways to heal tissue and fight off infections (Singh et al., 2019). But long-term inflammation can cause apoptosis, aberrant cell growth, and help tumors invade and grow new blood vessels (Chen et al., 2022; Tewari et al., 2021). TP stops the release of plasma inflammatory cytokines like TNF-α (Mao et al., 2023), IL-1β, IL-6 (Chen et al., 2021), MCP-1 (Huang et al., 2021), MMP-3, MMP-9 (Piao et al., 2021), Cox-2, and NLRP3 (Fang et al., 2021). TP has strong anti-inflammatory effects when it comes to inflammation in bones (Chen et al., 2021). After TP therapy, there were big drops in the number of F4/80 macrophages and CD3 T cells in inflammatory lesions (Li, Yang et al., 2020). If you have collagen-induced arthritis (CIA), TP’s antioxidant action could be useful for treatment (Yu et al., 2021).
Serum amyloid A (SAA) is an acute-phase protein that can build up in the wrong places and cause AA amyloidosis, which is a disorder that happens when chronic inflammatory diseases damage more than one organ (Jensen & Whitehead, 1998; Uhlar & Whitehead, 1999). TP lowers SAA levels, stops amyloid from building up, and helps the body get rid of it, which lowers the number of cases of amyloidosis in mouse models (Cui et al., 2007). TP also controls immune cells like T cells, B cells, and monocytes to stop inflammation and the release of cytokines (Chang et al., 1997; Qiu et al., 1999).
Studies in vitro on human NSCLC A549 cells show that TP and hydroxycamptothecin (HCPT) work together to increase PP2A activity by lowering the catalytic C subunit, raising the structural A subunit, and lowering phosphorylation at Tyr307 on PP2A-C (Turowski et al., 1997). This combination also activates ERK and p38-MAPK by targeting kinases that are higher up in the signaling pathway. This shows that it could be useful in cancer treatment.
TP Reverses Tumor Drug Resistance
Paclitaxel, cisplatin, and gefitinib are medications that are often used to treat NSCLC. However, as treatment goes on longer, they may become resistant to drugs, which makes them less useful in the clinic. Wang et al. (2015) found that tumor cells may employ the nucleotide excision repair (NER) pathway to fix DNA damage produced by platinum medicines. This makes the treatments less effective because they cannot get through the cell membrane as easily. TP can stop the ATPase activity of the TFIIH basic transcription factor by binding to it. TFIIH is also a part of the NER pathway. At a modest dosage (5 ng/mL), TP can stop NER activity and make cisplatin kill lung cancer cells more effectively. Studies have also demonstrated that TP stops the growth of drug-resistant A549 cell xenografts by increasing the expression of E-cadherin, which is linked to the reversal of EMT (Li, Cui et al., 2020). A critical part of the EMT process is the loss of E-cadherin expression, which is a sign of malignancy. Researchers (Jiang et al., 2016) also observed that TP can stop the production of NF-κB-controlled drug resistance genes FLIP, XIAP, Bcl-2, Bcl-xL, and COX-2 and make lung cancer less resistant to paclitaxel. FLIP and XIAP stop caspases from working, and having a lot of them can make tumors resistant to drugs. COX-2 can help blood vessels grow and stop the immune system from working (Table 1).
Targets and Methods of Triptolide (TP) in the Treatment of Lung Cancer.
Clinical Application of TP
A new study demonstrates that TP can efficiently go beyond NSCLC’s resistance to paclitaxel by blocking the HNF1A/SHH axis. Paclitaxel resistance is a big problem when treating NSCLC. TP makes paclitaxel-resistant cells more sensitive to paclitaxel by lowering the expression of ABCB1 (Li, Yang et al., 2024). TP had a good anti-tumor effect in both in vitro and in vivo studies. This opened up a new option for treating NSCLC patients who are resistant to paclitaxel. Researchers have come up with a number of medication delivery techniques to make TP more effective and less harmful for treating lung cancer. For instance, researchers used exosomes from milk to carry TP and then gave it to lung cancer models via mouth. The results indicated that the TP inside this exosome was more easily absorbed by the body and stopped lung cancer cells from growing significantly. When compared to free medicines, the rate of tumor inhibition went up by 25% (Sun et al., 2024). The researchers also used liposome technology to encapsulate TP, which made it easier for the intestines to absorb it (Sun et al., 2024). TP has also been found to work through a variety of mechanisms. For example, it can block the G1 phase process of lung cancer cells by inhibiting the expression of cyclin D1, thereby inhibiting cell proliferation (Xu et al., 2020). Researchers have also revealed that TP works in a number of ways. For instance, it can stop lung cancer cells from going through the G1 phase by stopping the expression of cyclin D1, which stops cells from growing.
Summary
This review goes into great detail about how triptolide and TP work on multiple targets to treat lung cancer and shows that they could be useful in the fight against lung cancer. This study systematically summarized how TP affects lung cancer cells in a number of ways, such as causing apoptosis, controlling the immune response, stopping invasion and migration, fighting oxidation, lowering inflammation, and reversing tumor drug resistance. This is different from what other studies have done. These mechanisms span several important steps in the growth of lung cancer and help us understand how TP fights cancer in a more complete way. Second, this study looked into how TP causes copper death in lung cancer cells for the first time. TP raised the amount of copper ions inside cells, which damaged the mitochondrial membrane potential and turned on the iron–sulfur cluster protein breakdown process. This eventually killed the cells. This discovery gives us a new way to think about how TP fights cancer and also gives us a theoretical framework for coming up with new ways to fight cancer that are based on copper death.
This study also looked at how TP affects the immunological escape of lung cancer cells when it is used as an intervention. TP may help DCs mature and become active, boost the immune system’s ability to fight tumors, and stop tumor cells from blocking the immune system by controlling the expression of immune checkpoint molecules like PD-1 and PD-L1. These results give us fresh ideas for how to use immune regulation to treat lung cancer. At the same time, research has shown that TP lowers oxidative stress damage by controlling the Nrf2/Keap1 signaling pathway and increasing the production of antioxidant enzymes like HO-1 and NQO1. TP also stops tumors from growing by stopping the release of several inflammatory factors (including TNF-α, IL-6, and MCP-1) and lowering the inflammatory response. This study also investigated how different drug delivery mechanisms, such as exosomes and liposomes, could be used to make TP more available and less harmful. These novel ways of delivering drugs have made TP far more effective in treating lung cancer. They are an excellent example for coming up with new ways to distribute drugs. In short, this study not only gives a systematic overview of the multi-target mechanism of TP in the treatment of lung cancer, but it also shows its new mechanisms in copper death, immune escape intervention, anti-oxidation and inflammation regulation, and reversal of tumor drug resistance. This is an important theoretical basis and experimental support for the development of new lung cancer treatment strategies based on TP. Future studies will look into how TP can be used in the clinic and provide lung cancer patients more hope.
Footnotes
Abbreviations
AMPK: AMP-activated protein kinase; CaMKKβ: Ca²+-dependent kinase; CIA: Collagen-induced arthritis; DCs: Dendritic cells; ERK1/2: Extracellular regulated protein kinase 1/2; HCPT: Hydroxycamptothecin; HSP70: Heat shock protein 70; IFN-γ: Interferon-γ; MAPK: Mitogen-activated protein kinase; MDM2: Mouse double minute gene; NER: Nucleotide excision repair; NSCLC: Non-small cell lung cancer; PD-1: Programmed death protein 1; PD-L1: Programmed death protein ligand 1; PI3K: Phosphatidylinositol 3-kinase; ROS: Reactive oxygen species; RPL23: Ribosomal protein L23; SAA: Serum amyloid A; SIRT3: Deacetylase 3; TNF-α: Tumor necrosis factor-α; TP: Triptolide.
Authors’ Contributions
Qin Dakai, Zhang Qinyuan, and Lin Demin designed the experiments; Yang Maoqin performed experiments and collected data; Qin Dakai, Zhang Qinyuan, Ren Dexiang, and Fu Yunyun discussed the results and strategy; Yang Maoqin and Fu Yunyun supervised, directed, and managed the study; Xia Xiaojun and Lei Dongxu final approved the version to be published.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical Approval and Informed Consent
Not applicable.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Gansu Provincial Administration of Traditional Chinese Medicine (Grant No. GZKZD-2018-03), the Natural Science Foundation of Gansu Province (Grant No. 23JRRA1252), the Gansu Provincial Famous Traditional Chinese Medicine Inheritance Studio Construction Project (Guozhong Medicine Gui Cai Han [2021] No. 242) and the Gansu Provincial Natural Science Foundation (Grant No. 23JRRA768).