
Abstract
The pathophysiology of dystonia in Wilson disease (WD) is complex and poorly understood. Copper accumulation in the basal ganglia, disrupts dopaminergic pathways, contributing to dystonia’s development via neurotransmitter imbalance. Despite advances in diagnosis and management, WD with dystonia remains a challenging condition to treat. We aim to report the unprecedented co-occurrence of pathogenic genetic variants in both the ATP7B and KMT2B genes in a patient with WD. A 13-year-old male presented at 12 with dysarthria and bilateral Kayser-Fleischer rings. Over months, dystonia spread to his left foot, upper limb, and trunk, accompanied by slowed daily activities. Diagnostic tests included MRI for brain structure, abdominal ultrasound for liver function, serum ceruloplasmin and copper levels to assess copper metabolism, and 24-hour urine copper tests for excretion levels. Whole exome sequencing was conducted using genomic DNA from peripheral blood samples. Variant classification followed guidelines from the American College of Medical Genetics and Genomics. The sequencing revealed compound heterozygous pathogenic variants in the ATP7B gene: NM_000053.4:c.2165dupT and NM_000053.4:c.813C>A. A pathogenic variant in the KMT2B gene, NM_014727:c.3052delA, was identified. This case highlights WD co-occurrence with ATP7B and KMT2B mutations, suggesting KMT2B as a potential genetic modifier.
Introduction
Wilson disease (WD) is a relatively rare condition, with an estimated prevalence of 1 in 30,000 to 1 in 50,000 individuals worldwide. 1 This disorder arises due to genetic mutations in the ATP7B gene, which result in the production of altered ATP7B protein. ATP7B plays a crucial role in regulating copper metabolism within the body. 2 The manifestations of WD can vary widely, affecting multiple organs and systems. The accumulation of copper primarily affects the liver and brain, leading to hepatic and neurological symptoms. Hepatic symptoms include hepatomegaly, jaundice, and liver dysfunction, while neurological symptoms may include movement disorders, psychiatric disturbances, and cognitive impairment.
In recent years, another rare movement disorder known as DYT-KMT2B has garnered attention. KMT2B dystonia, also referred to as dystonia 28 or DYT28, is an autosomal dominant neurological condition that manifests as early onset generalised dystonia with prominent cervical, oromandibular and laryngeal involvement. 3 The complex phenotype of DYT-KMT2B is characterised by dysmorphic features, intellectual disability, developmental delay, and microcephaly. ClinVar has reported 127 pathogenic or likely pathogenic variants in the KMT2B gene involved in dystonia. 4 However, no reported cases or evidence of co-occurrence of pathogenic variants in the ATP7B and KMT2B genes have been reported. We report a case of a 13-year-old boy with lingual dystonia and confirmed genetic findings of a pathogenic variant in the ATP7B gene and a co-occurring pathogenic variant in the KMT2B gene.
Approval was taken from the Ethical Review Committee of our institution and the patient’s identity has not been revealed. No photographs have been used. Understanding the genetic underpinnings of these disorders is crucial for accurate diagnosis, appropriate management, and potential future therapeutic interventions. Further investigations and comprehensive genetic studies are warranted to shed light on the complex genetic landscape and potential associations between WD and other movement disorders such as DYT-KMT2B.
Case Presentation
Index Case
A 13-year-old male child, firstborn of non-consanguineous parents, presented to us with dysarthria and lingual dystonia from 12 years of age. Over the next few months of onset, dystonia progressed to involve the left foot, upper limb, and subsequently the trunk, along with slowness in all activities of daily living. On examination, he had severe generalised dystonia and could ambulate with bilateral support. Kayser-Fleischer rings were present bilaterally. MRI brain revealed bilateral atrophy of the caudate nucleus, symmetrical T2 hyperintensity and central hypointensity in both putamina and cerebellar atrophy. Susceptibility weighted imaging (SWI) showed areas of blooming in both putamina, globus pallidus and substantia nigra (Figure 1). Ultrasonography of the abdomen showed an altered liver echotexture. Biochemical and radiological evaluations (Table 1) were consistent with WD. Family history is depicted in Supplementary Figure 1.
Biochemical and Radiological Investigations.
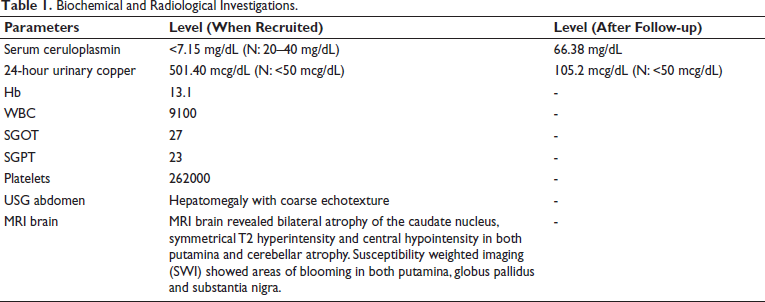
MRI Images Showing Implications Associated with the Brain. (a) Bilateral atrophy of the caudate nucleus is observed, with symmetrical T2 hyperintensity and central hypointensity noted in both putamina. (b) Mild diffuse cerebellar atrophy is evident, characterised by prominent cerebellar folia. (c and d) Coronal T2-weighted imaging reveals bilateral caudate nucleus atrophy. (e and f) Susceptibility Weighted Imaging (SWI) depicts the flourishing of putamen, pallidum, and substantia nigra.
Genetic Analysis
Whole exome sequencing was performed on the Illumina platform. The sequencing protocol began with the isolation of genomic DNA from the blood samples, followed by library preparation using the Twist Comprehensive Kit. Paired-end libraries were generated after gDNA fragmentation (150–200 base pairs). To capture the exonic regions (coding region) 100 ng of fragmented gDNA were incubated with biotinylated RNA capture baits. Further, the captured fragments were sequenced on NovaSeq with 150 paired-end reads. The quality of the reads was checked by FastQC and Trimmomatic was used for trimming, filtering and adapter removal. Sequencing reads were mapped against the human reference genome (GRCh38) using variant calling performed on the GATK pipeline and vcf files for all samples were combined using bcftools. Variant annotation was done on ANNOVAR. Variants were filtered based on minor allele frequency (MAF <5%) from different population datasets like 1000 genome, ExAC and ESP6500. We prioritised variants in dystonia-related and liver disease-related genes and the variants were classified according to the ACMG-AMP guidelines.
Genetic Findings in the Patient
We identified pathogenic genetic variants in both the ATP7B and KMT2B genes. In the ATP7B gene, two compound heterozygous variants, NM_000053.4:c.2165dupT (exon 8) and NM_000053.4:c.813C>A (exon 2) were detected and classified as pathogenic. For the KMT2B gene, the variant NM_014727:c.3052delA was found in exon 7 and assigned the pathogenic classification based on the ACMG-AMP guidelines. The IGV (Integrative Genomics Viewer) of variants is depicted in Figure 2.
Integrative Genomic Viewer (IGV) Accentuating Compound Heterozygous Variants in ATP7B (p.C271X/p.R723Efs*31) and a Heterozygous Variant in KMT2B (p.G1020Afs*3).
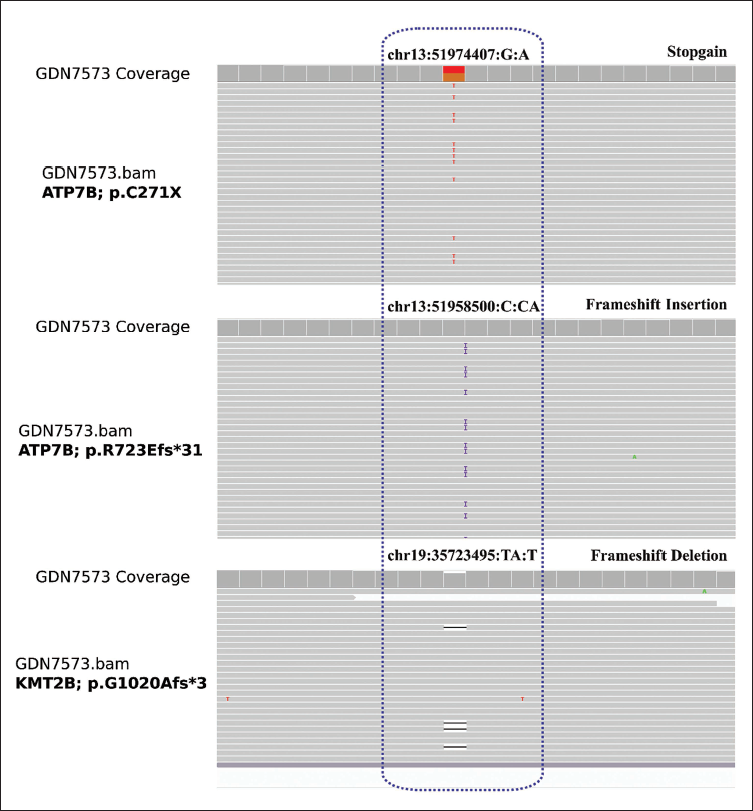
To classify the genetic variant p.G1020Afs*3 (NM_014727:c.3052delA) in the KMT2B gene, we applied the ACMG-AMP guidelines. Multiple criteria were considered in the classification process. PVS1 (loss of function) and PM2 (low minor allele frequency in 1000GP, ESP6500, and ExAC) were applied. Additionally, PP3 (at least two out of three in silico tools indicating harmful impact on the protein; CADD, PolyPhen2_HVAR, and SIFT) were taken into account. The combination of these attributes led to the determination of the pathogenic nature of the variant.
Treatment and Outcomes
The patient received treatment for WD with copper chelators (D-penicillamine and Zinc) along with symptomatic medical management for dystonia (Trihexyphenidyl, Baclofen, Benzodiazepines). At a 5-year follow-up, he has significant clinical improvement- with a reduction in dystonia and regaining ambulation without support.
Discussion
In this report, we underscore the rare co-occurrence of pathogenic variants in both the ATP7B and KMT2B genes within a single individual. Within the ATP7B gene, two compound heterozygous variants, namely p.C271X and p.R723Efs*31, were identified. These variants have previously been classified as deleterious, 5 further supporting their association with WD. Additionally, a specific pathogenic variant, p.G1020Afs3, was identified in the DYT-KMT2B gene. This variant is a frameshift deletion that has been previously described in the literature as causing loss of function. Based on this information, it was classified as a pathogenic variation, indicating its detrimental impact on the function of the gene. Although dystonia is a shared symptom in both diseases, clinical findings and response to treatment were most consistent with WD in our patient.
The unique co-occurrence of ATP7B and KMT2B variants may have several implications. Given the clinical phenotype strongly consistent with WD, it may be considered whether the KMT2B variant is an incidental finding that is not relevant clinically. However, a stringent interpretation of the ACMG-AMP guidelines and prior reports of this specific variant in patients with DYT-KMT2B suggest that this is unlikely to be functionally irrelevant. It may be hypothesised that the KMT2B variant has an effect modifier role on the clinical phenotype of WD in this patient. Indeed, genetic variations in additional genes may play a role in the manifestations, such as age at onset, clinical symptoms and response to therapy. There are also reports of patients with WD, who have suboptimal response to treatment, or worsen with copper chelators and respond to Deep Brain Stimulation as a symptomatic therapy. 6 The role of tandem variants such as ATP7B/KMT2B needs to be further explored in such subgroups.
These genetic findings shed light on the complex genetic background underlying movement disorders in an individual patient. This study expands our understanding of the genetic mechanisms involved in movement disorders and emphasises the importance of considering multiple genetic factors when evaluating and managing patients. Further studies and investigations are warranted to elucidate the precise interactions and underlying molecular pathways that contribute to the development of these complex conditions. Such research efforts will be instrumental in advancing our knowledge and facilitating the development of more effective diagnostic and therapeutic approaches for individuals affected by WD and DYT28 dystonia.
Footnotes
Acknowledgement
MK acknowledges the support of a Senior Research Fellowship from CSIR. Additionally, we extend our gratitude to Dr. Karthik for his contribution to the MRI acquisition and interpretation.
Authors’ Contribution
BBK conceptualized and supervised the analysis. MK performed data analysis, variant interpretation and case report writing. AA, AS, DMR and RR performed clinical interpretation and provided patient sample. SSB and VS provided whole exome sequencing data.
Statement of Ethics and Patient Consent
The study was approved by the Council of Scientific & Industrial Research (CSIR)-Institute of Genomics and Integrative Biology (IGIB), New Delhi, India, and the respective clinical centres (Ref no: CSIR/IHEC/2017-18, CSIR/IHEC/2018-19/IndiGen, CSIR/IHEC/2018-2020/RareGen).
The authors attest that they got all the necessary patient consent papers. The patient has provided his agreement in the form for his clinical pieces of information to be published in the journal. The patients understand that his name and initials will not be published.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Funding
The authors received funding support from the Council of Scientific and Industrial Research (CSIR; Grants MLP2008).
Supplemental Material
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.