
Abstract
Background
Parkinson’s disease (PD) is characterized by dopaminergic (DA) neuron loss, Lewy body build-up, and motor dysfunction. One of the primary pathogenic mechanisms of PD development is autophagy dysfunction and nitric oxide-mediated neurotoxicity.
Purpose
The current study focuses on autophagy and nitric oxide (NO) signaling roles in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-intoxicated PD mice and their protection by their modulators.
Method
BALB/c mice were administered MPTP (30 mg/kg/i.p/day) for five consecutive days in order to create a PD model. Following MPTP poisoning, the doses of GA (16.8 mg/kg/day/i.p.), 7-nitroindazole (7-NI) (10 mg/kg/day/i.p.), and their combination were administered once daily for 14 days. Animals were observed for behavioral and locomotor changes, biochemical examination, inflammatory mediators, and analysis of molecular markers.
Results
GA, 7-NI alone significantly reduced MPTP-induced locomotor, behavioral, and oxidative damage. Additionally, in MPTP-intoxicated animals, 7-NI and GA had protective effects on dopamine levels, TH positive DA neurons, inflammatory cytokines interleukin 1β (IL-1β), tumor necrosis factor-alpha (TNF-α), nuclear factor-kappa B (NF-κB), and cyclooxygenase-2 (Cox-2) concentration. Furthermore, GA increases LC3BII expression, which in turn increases autophagy. It also decreases total NO content, and a significant response of 7-NI demonstrates their interaction, which is neuroprotective.
Conclusion
Present research suggests that dysregulation of autophagy and NO-mediated neuroinflammation are involved in the pathogenesis and progression of MPTP-induced PD. The use of two pharmacotherapeutics, GA and 7-NI, respectively, significantly reduces MPTP-induced PD distortions and their interaction enhances the overall protective effect, suggesting that these pharmacological agents may be used for the treatment of PD.
Introduction
Parkinson’s disease (PD), with its distinctive Lewy body formation and dopaminergic (DA) neuronal cell death, is the second-most prevalent neurodegenerative disorder and the most prevalent movement disorder in the elderly population globally.1, 2
Previous studies have demonstrated that autophagy is a crucial pathway for α-Synuclein breakdown and may have therapeutic effects for individuals with PD. 3 Effective autophagy/mitophagy activation in response to mitochondrial damage or neuronal damage has a clear pro-survival role, and the process has been reported as being downregulated in certain models of PD.4–7 Among them, mitochondrial dysfunction appears to be of critical importance in PD because it can result in excessive reactive oxygen species (ROS) production, an inflammatory response, and the activation of cell death pathways.8, 9
Glycyrrhizic acid (GA), obtained from Licorice root (Glycyrrhiza glabra L.), a triterpenoid saponin is one of the oldest ayurvedic pharmacoagent. Numerous pharmacological characteristics of GA, including anti-inflammatory, antioxidant, antiviral, anticancer, gastroprotective, cardioprotective, hepatoprotective, and neuroprotective effects, have been demonstrated. 10 The neuroprotective potential of GA in research models of cerebral, spinal, and ischemic brain injury has recently gained interest. According to certain research, GA significantly protects neurons from damage induced by 6-hydroxydopamine (6-OHDA) or glutamate.11, 12 Although less is known about GA’s efficacy in vivo models, recent researches show that it can activate autophagy in diverse cell lines for cancer and neurological disorders in a concentration-dependent manner.13, 14
Moreover, increasing data indicate that excess production of nitric oxide (NO) is implicated in aging processes and the development of PD.15, 16 It has been demonstrated that excessive NO generation causes damage to neurons and is linked to the S-nitrosylation or nitration of various crucial proteins, such as Parkin, protein-disulfide isomerase, mitochondrial complex I, peroxiredoxin-2, and α-synuclein in the PD neurodegenerative process. Patients with PD and animal models of the disease show high levels of neuronal NOS (nNOS) and inducible NOS (iNOS) in the substantia nigra (SN). 17 An NOS inhibitor that targets neuronal isoforms, 7-nitroindazole (7-NI), demonstrated neuroprotective effects against 6-OHDA and 1-methyl-4-phenylpyridinium (MPP+)-mediated toxicity.18–23
Furthermore, different neurodegeneration models show increased NO inhibition of autophagy, potentially leading to increased cellular stress. Increased NO production may harm cells via oxidative or nitrosative stress.24, 25 Moreover, recent research also showed that nitric oxide and autophagy interact somewhat and that ROS-mediated nitrosative stress and autophagy dysregulation may play a role in the etiology of neurological diseases. 26 According to a recent study of Sarkar, 27 NO can inhibit JNK1 (c-Jun N-Terminal Protein Kinase1) and IKK (I Kappa-B Kinase) via mTOR (mammalian target of rapamycin) and mTOR-independent pathways, respectively, to prevent autophagy. One more study shows the inhibition of autophagy by nitric oxide during hypoxia-reoxygenation may contribute to bioenergetics impairment and promote cell death in neurons. 28 Hence, increasing autophagy while decreasing NO may be an additional therapeutic benefit in order to maintain a healthy lifestyle and limit the progression of the disease in PD patients.
Therefore, in our investigation, we attempted to comprehend the interaction between autophagy and nitric oxide signaling in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-intoxicated animals. We found that the combination of an autophagy inducer and an NOS inhibitor provided the significant protection against MPTP neurotoxicity.
Material and Methods
Experimental Animals
The male BALB/c mice were purchased by the CSIR-IITR in Lucknow when they were 18–20 weeks old and 30–35 g. The institutional animal facility at the university offered an aseptic environment for the experimental animals, which had a temperature of 23°C–24°C, a humidity of 50%–70% and a cycle of 12 h of light and 12 h of darkness, with free access to food and drink. All of the experiments received approval from the King George’s Medical University (KGMU), Lucknow’s Institutional Animal Ethics Committee (project 88/IAEC/2017).
Experimental Design
All of the study’s experimental animals are split into the following groups (N = 8): Group I received only saline; and Group II received dose of MPTP (30 mg/kg/i.p/day) for five consecutive days. 29 Freshly prepared GA (16.8 mg/kg/day/i.p.) diluted in normal saline was given to Group III, whereas Groups IV were treated with 7-NI (10 mg/kg/day/i.p.) dissolved in peanut oil according to earlier studies.18, 25, 30, 31, Groups V and VI were given MPTP+GA and MPTP+7-NI, respectively, after 1 h of MPTP induction on day one and continued for 14-day schedule. Group VII was used to assess the additive effects of GA and 7-NI in MPTP-intoxicated mice. Every drug is administered in accordance with earlier literatures and is then further standardized.
We studied mouse behavior and locomotion to determine the degree of Parkinsonism. After that, four animals from all experimental groups were killed by cervical dislocation while under anesthesia, and their intact brains were removed, and four animals from each group were subjected to cardiac perfusion with 4% paraformaldehyde and fixed for immunofluorescence analysis. These were then sectioned into the SN and striatum and used for biochemical, neurochemical, and immunofluorescence analyses.
Reagents and Antibodies
MPTP hydrochloride (23007-85-4), 7-NI (2942-42-9) RIPA lysis buffer (632424), protease and phosphatase inhibitors (P-8340 and P0044), and dithiothreitol (DTT) (10197777) were purchased from Sigma-Aldrich, St. Louis, MO, USA. GA (G0150) was purchased from Shanghai TCI Chemical Industry Co., and Bradford protein estimation kit (2603300011730) was purchased from GeNei™, Bangalore, India.
Rabbit polyclonal anti-LC3B antibody (ab48394) was purchased from Abcam, MA. Rabbit anti-human TH antibody (PAB438Bo01) and HRP-linked goat anti-rabbit IgG Ab (SAA544Rb19) were procured from cloud clone corp., USA. Antifading mounting media with DAPI (ENZ-53003-M010; Enzo Life Sciences, Farmingdale, NY) were procured. Alexa Fluor® 594 goat anti-rabbit IgG red (A11012), Griess reagent kit (G7921), TNF-α Mouse (BMS607HS), and IL-1β Mouse (BMS6002) enzyme-linked immunosorbent assay (ELISA) kits were procured from Thermo Fisher Scientific, Waltham, MA. Mouse nuclear factor-kappa B (NF-κB) (SEB824Mu) ELISA Kit was purchased from cloud clone corp., USA. Mouse cyclooxygenase-2 (COX-2) (CSB-E12910m) ELISA Kit procured from Cusabio Houston, TX 77054, USA.
Behavioral Analysis
Actimeter
Using an actimeter (Columbus, Ohio, USA) and Opto Varimax-5 Auto-Track software, many behavioral changes in freely moving mice were measured. The experiment was conducted in accordance with earlier studies.32–34 Mice were subjected to a 5-min practice session afterwards, and each mouse of a different group was placed at the center of the arena (17.5 by 17.5) and allowed to move for 5 min. The experiment was designed to quantify locomotor activity, ataxia, and stereotyped behavior in terms of ambulation, speed, and the rearing parameter.
Rotarod Test
Mice were subjected to a 5-min practice session on a rotarod apparatus at a fixed or accelerating speed (0–30 rpm) to evaluate motor coordination in accordance with previous research.35, 36 After then, the animals in all of the experimental groups were watched for latency to fall at various rates of 15 and 30 rpm
Grip Strength Test
According to Brooks et al. technique, 37 the grip strength meter (Columbus Company, USA) was used to measure the maximum force displayed in order to assess neuromuscular functioning. To measure factors such as forelimb strength and total strength in units of Newton, mice from each group were subjected to a test grid (N) in order to measure the force shown on the transducer.
Neurochemical Analysis
HPLC Analysis
According to Liu J. Study, 38 we use reverse-phase HPLC (Agilent 1260 Infinity Quaternary HPLC) with a suitable diode-array detector to measure the amount of dopamine in the striatum of mice brains. The striatum tissue from the brain was removed, weighed, and then homogenized in 0.1-mol/l perchloric acid. The supernatant material was exposed to an HPLC column containing mobile phase under suitable analytical conditions following centrifugation at 13,000 rpm for 15 min at 4°C. The amount of dopamine (DA) in the tissue was quantified and expressed in ng per mg of wet tissue.
Estimation of Lipid Peroxidation
Ohkawa et al. 39 established a method for measuring lipid peroxidation. After homogenizing and measuring brain tissues for Bradford assay protein quantification, thiobarbituric acid (TBA) was quantified. Further, 10% SDS solution was added to 0.1 ml of 10% tissue homogenate before being incubated at room temperature for 5 min. The sample was treated with 0.6 ml of 20% acetic acid for 2–5 min, and then 0.6 ml of 0.8% TBA for 1 h at 65°C in a water bath to produce the recognizable pink color. The samples were settled, chilled, and then centrifuged. The malondialdehyde (MDA) concentration was measured in relation to a blank using spectrophotometry at 532 nm.
Superoxide Dismutase (SOD) Activity Measurement
The Marklund and Marklund technique was utilized to evaluate SOD activity in mouse brain tissue homogenates, 40 and the results were expressed in units per gm of protein. Each group’s brain samples were taken, homogenized, and then treated with pH 8.2 Tris-EDTA buffers and 0.2 mM pyrogallol solution. SOD was quantified in terms of content of present or absent while measuring the rate of autoxidation at 420 nm.
Catalase Activity Measurement
The catalase experiment was performed on mouse brain tissue homogenate, which had been prepared after the mice were sacrificed under anesthesia. After each sample was treated with H2O2 for a different amount of time, the residual H2O2 was measured. 41 The rate of H2O2 disappearance is measured in U/gm of protein and expressed as a drop in absorbance at 240 nm.
Proinflammatory Cytokine Estimation
Following the manufacturer’s instructions, TNF-α and IL-1β cytokines were quantified using an ELISA (Thermo Fisher Scientific, Waltham, MA). The euthenics measure was applied in processing of brain samples. Before centrifugation at 14,000g for 20 min at 4°C, SNpc of brain tissue was homogenized in assay buffer. All experimental groups’ samples were passed through ELISA strips coated with the appropriate antibody before being evaluated in accordance with the kit’s methodology using detection limits for TNF-α and IL-1β of 5 and 25 pg/ml, respectively.
NF-κB Estimation
At the end of the study, mice were anaesthetized and executed; their brains were then taken and processed in 10% phosphate buffer saline (PBS). After that, using a commercial ELISA kit, NF-κB expression in brain lysate was measured in terms of pg/g (Cloud Clone Corp.).
Cox-2 Activity Estimation
According to protocol of Cox-2 ELISA kit from CUSABIO and our earlier studies, 32 the activity of the Cox-2 was assessed in mice brain tissue homogenates and expressed as pg/g tissue.
Immunofluorescence
During the immunofluorescence investigation, animals were anaesthetized, transcardially perfused, and then preserved with 4% paraformaldehyde. Brains were cut into 1–3 µm coronal sections and placed on PLL-coated slides using a cryomicrotome (Cryostat, CM1950, Leica Biosystems, Germany). Slides were cleaned in PBS, treated with citrate buffer for antigen retrieval, blocked with 3% horse serum in BSA for 1 h, and then incubated overnight with primary antibodies in (1:100) concentration, such as rabbit anti-human TH antibody and polyclonal anti-LC3B antibody. Afterward, Alexa Fluor fluorescent secondary antibodies (1:200) (Red 546 nm) were incubated for 1 h in the dark, treated with antifading mounting solution with DAPI, examined in a fluorescence microscope (Olympus BX53F2, Tokyo, Japan), and analyzed using Image J software developed by the National Institutes of Health.
Statistical Analysis
All statistical analyses were carried out using GraphPad software (Inc.; version 6). Results were expressed as means ± SD, and statistical significance was established using the one-way and two-way ANOVA followed by the Neumen-keul post hoc test for all. p-values <.05 were considered statistically significant.
Results
Effect of GA and 7-NI on Motor Impairment and Neuro Behavior Parameter in MPTP-induced PD Mice
Different parameters were evaluated in order to evaluate the locomotor and neuromuscular alterations in MPTP-intoxicated mice and their protection by GA and 7-NI alone and in combination. We examine the ambulatory, rearing, and speed parameters in mice after 14 days of treatment using an activity meter (OptoVarimex 5, Columbus Instruments, USA). MPTP induction greatly reduces the ambulatory, rearing, and speed responses associated with Parkinsonism (Figure 1a), but treatment with GA and 7-NI dramatically improves these MPTP-induced reductions. Additionally, we discovered that co-treating mice with GA and 7-NI was almost equal to mice in the control group, proving the additive protective potential of GA and 7-NI.
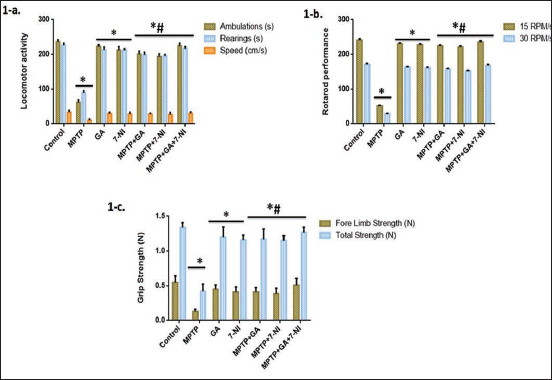
We then assess motor dysfunction and neuromuscular strength across distinct groups. After a 14-day treatment period, the mice’s grip strength in terms of forelimb strength and total strength was assessed using the Grip Strength Meter (Columbus Instruments, USA) (Figure 1c). Mice’s grip strength significantly decreased after the administration of MPTP. Compared to MPTP-induced mice, regular treatment with GA and 7-NI protects motor behavior, with the combined effect being the most significant.
Next, following a training session at a fixed or accelerating speed, mice were subjected to rotarod apparatus on various rotation schedules at a 15 and 30 rpm, and their latency to fall was recorded 14 days after MPTP induction. The locomotive power of mice significantly decreased following MPTP intoxication. Even though GA and 7-NI alone have a conventional tendency similar to control, when combined with MPTP, it considerably increases locomotor strength with the most remarkable effect in synergism (Figure 1b).
Effect of GA and 7-NI on Oxidative Damage in MPTP-induced PD Mice
Mitochondrial dysfunction has a significant impact on the etiology of Parkinson’s disease and the inflammatory responses brought on by oxidative stress. Consequently, we found in our research that MPTP, a mitochondrial neurotoxin, effectively elevated the degree of lipid peroxidation (MDA) and impaired the antioxidant system. We discover that GA and 7-NI alone have nearly same MDA levels as in controls, and when administered to MPTP-intoxicated mice, they successfully achieve a primary neuroprotective goal by bringing the level down to close to normal, which shows the anti-oxidative effect of both drugs (Figure 2a).
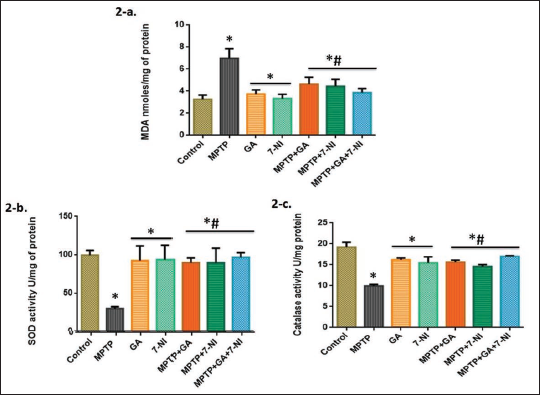
In context to evaluate the antioxidant capacity of GA and 7-NI in MPTP-induced mice, SOD and Catalase were examined. Mice with MPTP induction displayed a marked decline in SOD and Catalase activities. Treatment with GA and 7-NI both alone and in combination induces a protective response and causes the concentrations of the aforementioned antioxidant enzymes to rise noticeably (Figure 2b and c).
Effect of GA and 7-NI on Inflammatory Markers and Dopamine Level, in MPTP-induced PD Mice
According to previous studies, the fundamental cause of PD may be neuroinflammation.42, 43 To assess the inflammatory consequences of MPTP intoxication, we use ELISA kits to measure the levels of cytokines such as TNF-α and IL-1β (Figure 3a and b), as well as the concentrations of NF-κB and Cox-2 activity (Figure 4a and b). We found that MPTP administration shows a marked increase in these markers’ levels, whereas alone treatment of GA and 7-NI was almost consistent with control. Moreover, with optimal protection in synergisms, the anti-inflammatory drugs GA and 7-NI successfully ameliorate MPTP-induced changes.
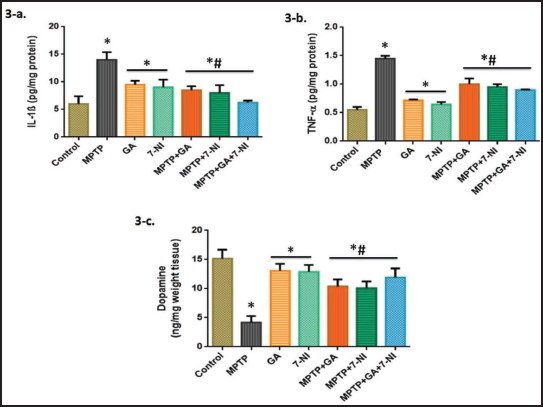
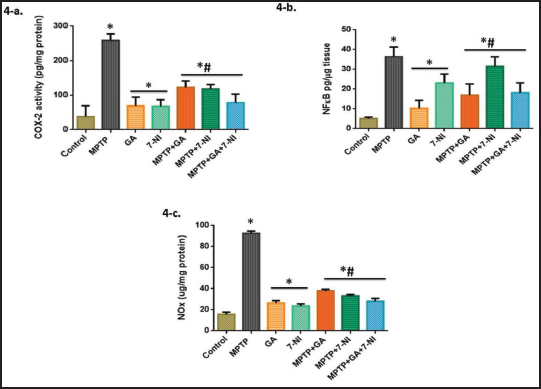
Following that, we used HPLC analysis to determine the characteristic dopamine level in mice striatum. We discovered that MPTP induction markedly reduces dopamine content, while GA and 7-NI alone and in MPTP intoxication significantly increase the level. However, co-treating GA and 7-NI did not demonstrate a significant additive effect and was similar to alone treatments (Figure 3c).
Amelioration of Impairment of Autophagy and Nitric Oxide Signaling in MPTP-induced PD Model
Our study emphasizes the dysregulation of autophagy and nitric oxide signaling and their interaction in MPTP-induced PD mice model.
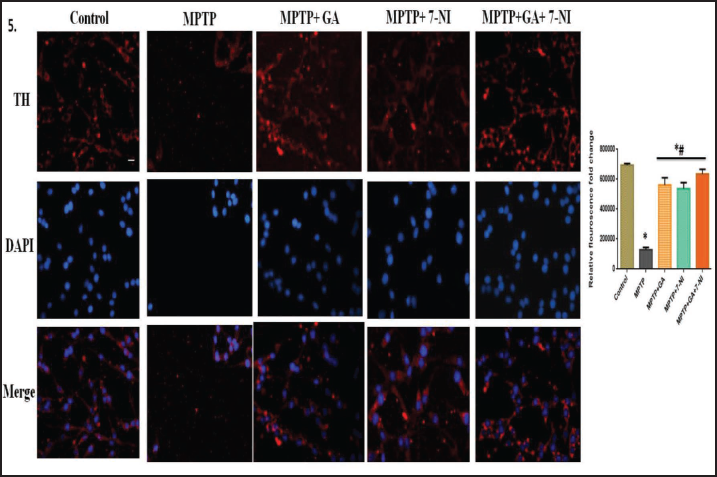
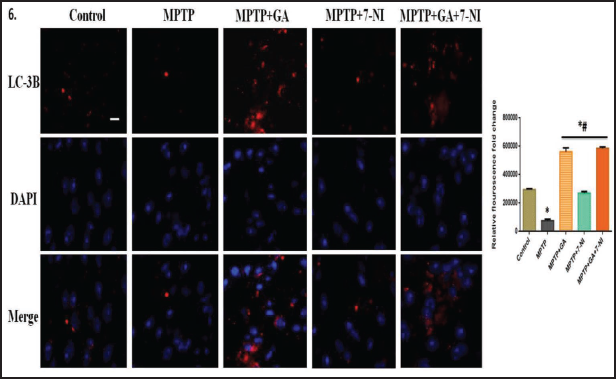
Characteristic marker proteins were evaluated to see whether the aforementioned pathways were involved. When determining the number of TH+ neurons per SN in the determination of DA neurons, we used immunofluorescence detection on mouse brain tissue sections. We discovered that TH+ immunoreactivity was significantly decreased in MPTP-treated mice when compared to the control, and that treatment with GA and 7-NI significantly reversed these changes (Figure 5). Moreover, we look at LC3BII, a hallmark autophagy protein to evaluate the damage induced by MPTP and the protection provided by GA. In line with the conventional theory, which holds that increasing autophagy will reverse the pathogenic changes in PD, our pharmacological agent GA successfully illustrates the same whereas 7-NI has less significant response (Figure 6). Additionally, we measure the total NO activity in several treatment groups in order to evaluate the MPTP-induced changes in nitric oxide level using the Griess reagent, and we discover that not only 7-NI, but GA also noticeably eliminates the MPTP-intoxicated changes (Figure 4c).
Discussion
The current pharmacotherapies primarily exhibit refractory effects, which are dependent on dopamine restoration, and are symptomatic in nature. They are also not useful for long-term persistence of disease development. The challenges of late-stage problems highlighted the importance of developing and implementing neuroprotective strategies that may be applied in the early stages and aid in preventing disease outcomes.
In context to this, in the present study, we used two neuroprotective agents such as GA, an autophagy inducer, and NOS inhibitor 7-NI and attempted to establish the relationship between autophagy and nitric oxide signaling. We find that these two pathways are interconnected with one another and with Parkinson’s pathogenesis, and by targeting these pathways, we will able to restore the underlying pathogenesis of PD.
We selected the mitochondrial neurotoxin MPTP in this study because it closely replicate the biochemical and nigrostriatal pathology seen in Parkinson’s disease and serves as a useful model for analyzing the PD pathology.1, 44 Our results confirm previous research that has showed that ROS generation and mitochondrial damage lead to inflammation 45 by demonstrating that MPTP intoxication modulate autophagy and over-expressed the oxidative and inflammatory markers in mouse brains.
Numerous studies suggest that nitric oxide may potentially play a role in MPTP neurotoxicity since inhibiting NOS attenuated the dopamine depletion caused by MPTP.20, 46, 47 Our findings support that MPTP-induced damage drastically reduces the dopamine level and TH positive neurons in the SN of mice brains, both of which are characteristically low in PD.25, 48 We found that not only 7-NI but also GA alone and in their combination actively replenish the amount of dopamine and TH+ neurons in the SN and significantly reduce the MPTP-driven load in mice (Figures 3c and 5).
We investigated that MPTP therapy increases lipid peroxidation while decreasing locomotor behavior and antioxidant enzymes. GA has recently been found to improve motor performance in the lipopolysaccharide and rotenone intoxicated PD mouse model,10, 49 whereas 7-NI has long been neuroprotective outcomes.19, 46 Data from different evaluations show that GA and 7-NI alone significantly recovered the MPTP-induced changes in behavior and cognitive impairment (actimeter test) and motor coordination (rotarod and grip strength test), while combined therapy considerably restored the MPTP-induced abnormalities to levels found in control mice (Figure 1). These results imply that combination therapy may have a significant therapeutic impact in PD neurobehavioral alterations suppression.
Additionally, MPTP poisoning results in a perceivable increase in MDA levels and a decrease in SOD and catalase levels, which is consistent with earlier studies that show MPTP to have an oxidative role in inducing DA auto-oxidation, enhancing basal production of hydrogen peroxide, and depleting antioxidant enzymes. 50 Our results demonstrate that both GA and 7-NI have protective effects, and interestingly, combination therapy successfully reverses the MPTP-induced alterations in oxidative damage by enhancement of antioxidant enzymes (SOD and catalase) and significant reduction in lipid peroxidation (Figure 2a and c). In addition, earlier neurodegenerative PD models exhibit elevated ROS levels, reduced expression of endogenous antioxidant systems, and mitochondrial dysfunction brought on by oxidative stress. 51 Together, GA and 7-NI have an inhibitory effect on the oxidative stress pathway, and their combination significantly minimizes the burden of oxidative stress in PD.
Studies have demonstrated that a rise in NO caused by ROS and nNOS has inflammatory effects and increase oxidant stress in PD.52, 53 One of the harmful outcomes of PD pathogenesis is an increase in pro-inflammatory cytokines, 54 and in this research, we assessed the key proinflammatory markers like TNF-α and IL-1β. Furthermore, we examined the impact of the principal inflammatory markers NF-κB and Cox-2 on mice’s brain, and we saw a notable enhancement in the MPTP-induced animals. Recent study predicted the inhibitory role of GA on NF-κB and Cox-2 levels10, 55 and we tried to extrapolate this in our MPTP animal model of PD. Interestingly, both GA and 7-NI exhibited a considerable decrease in the levels of these proteins, demonstrating their antioxidant and anti-inflammatory effects (Figures 3a, b and 4a, b). Additionally, the combination of GA and 7-NI exhibits cumulative effects, supporting the idea that the therapeutic index is increased when two anti-inflammatory drugs are added.
The connection between autophagy and NO was evaluated in MPTP-toxicated PD mice, and the hypothesis was supported by data from an assessment of number of parameters. The role of nitric oxide is well established in the MPTP model of neurotoxicity23, 25, 56 and in our research, we also observed that MPTP-instigation often raises NO levels (Figure 4c), whereas 7-NI, a specific NOS inhibitor, considerably lowers NO levels in PD mice consistent to our previous reports. 18 Surprisingly, we also discover that GA has a protective effect on NO activity, supporting the earlier findings27, 28 and our hypothesis that a decrease in NO signaling has a correlation with the activation of autophagy and that the two are partially related. However, more research is required to confirm this. Additionally, we attempted to look into the relationship between autophagy and PD, and the important marker protein of autophagosome formation LC3BII was assessed using immunofluorescence in the SN of mouse brain cortex. According to the data, there was a noticeable decrease in LC3BII expression after MPTP administration consistent with earlier reports.57, 58 In lung injury, the rotenone-mouse model and neuroblastoma cells, GA has been found to lessen the inflammatory effects and increase autophagy by the PI3K/AKT/mTOR pathway.10, 14, 59 Our findings corroborate these findings and GA was able to successfully reverse these alterations, whereas 7-NI alone or in combination had a less pronounced effect (Figure 6). Together, these findings have demonstrated that there is a connection between autophagy and NO signaling and that therapeutic success can be increased by focusing on them. In addition, more research is required to determine the relationship between autophagy and NO intermediaries and their therapeutic potential.
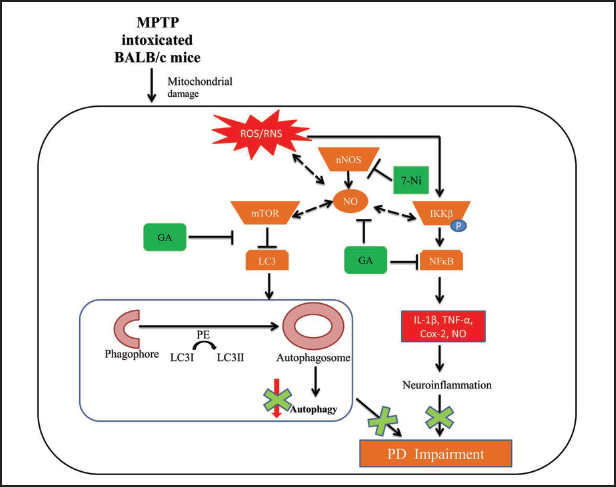
Consequently, our study shows (summary in Figure 7) that autophagy and NO signaling have vital role in MPTP-induced neurotoxicity, and that the protection provided by their modulators (GA, 7-NI, and their pharmaco-synergy) may be beneficial for treating Parkinson’s disease.
Conclusion
In summary, our research concludes the possibility that dysregulation in autophagy and NO plays a role in neuroinflammatory signaling and MPTP-induced damage. In this study, MPTP-intoxicated mice’s levels of inflammatory mediators, oxidative stress indicators, and reduced autophagy were all dramatically rescued while being protected from MPTP-driven DA degeneration, neurobehavioral abnormalities, and locomotor changes, which show that the synergistic effect of GA and 7-NI may have therapeutic effects on PD pathogenesis. Accordingly, we can conclude from our findings that these medicines have the potential to be employed therapeutically in the future to improve the quality of life for PD patients.
Footnotes
Abbreviations
Cox-2, Cyclooxygenase-2
GA, Glycyrrhizic acid
LC3B, Microtubule-associated protein-1 light chain 3B
MPTP, 1-Methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine hydrochloride
NO, Nitric oxide
7-NI, 7-Nitroindazole
NF-κB, Nuclear factor-kappa B
PD, Parkinson disease
ROS, Reactive oxygen species
SNpc, Substantia nigra pars compacta
TNF-α, Tumor necrosis factor-alpha
TH, Tyrosine hydroxylase.
Authors’ Contribution
The experiment was devised by Ms. Shipra Kartik and Dr. Rishi Pal.
Under the direction of Dr. Rishi Pal, Ms. Shipra Kartik conducted all of the tests and prepared the manuscript with assistance from Dr. Manju J. Chaudhary.
Dr. Madhu Kumar assisted with the pathological analysis and Dr. Rajendra Nath provided the technical advice.
Statement of Ethics
All of the experiments were granted permission by the King George’s Medical University’s (KGMU) Institutional Animal Ethics Committee (project 88/IAEC/2017).
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The Indian Council of Medical Research (ICMR), New Delhi, India awarded Ms. Shipra Kartik a senior research fellowship (Project-3/1/2/99/Neuro/2018-NCD-I).
ICMJE Statement
Form of ICMJE disclosure is attached.