
Abstract
Background
L1CAM protein plays a crucial role during early development and mutations in L1CAM cause L1 syndrome. L1 syndrome demonstrates a highly variable presentation within and between families. The clinical symptoms of L1 syndrome include mental retardation, hydrocephalus, spasticity, aphasia, and adducted thumb. Mutations in L1CAM gene were found to affect structurally essential key residues in extracellular region of L1 leading to changes in protein binding properties. In most cases, these mutations create unexpected phenotypes which need to be understood thoroughly.
Purpose
The L1 syndrome patients were identified by various phenotypes like mental retardation, hydrocephalus, aphasia, spasticity, adducted thumb, etc., and the patients or mental retardation (MR) children who had more than three symptoms. This study aimed to screen mutations in multiple exons by Sanger sequencing.
Methods
The present study employed primers which are designed for specific exons of L1CAM gene to amplify and sequence the amplified product to detect the mutations in L1 syndrome patients by the Sanger sequencing. Chi-square test was used to determine the mutation detection rate with the number of L1 syndrome phenotypes and several in silico programs were used to investigate potential effects of the variants.
Results
The nine different mutations in six patients. The mutation detection rate was high (83.33%) in patients with more than one L1 syndrome phenotype and in patients with more than one affected member in a family compared to patients with single phenotypes and negative family history (16.6%).
Conclusion
The mutation detection rate was related to the presence of typical L1 syndrome phenotypes and the family history. Screening of L1CAM gene mutations in the Indian population is much needed to analyze the mutations and understand the mechanism underlying L1 disease. The present study has identified some novel mutations which are implicated in alterations in various biological functions during development leading to pathogenesis of L1 syndrome.
Introduction
The human L1CAM gene encodes for the neural cell adhesion molecule L1 which is required for the development of the central nervous system. It mediates a wide range of CNS maturation-related activities, including neurite outgrowth, adhesion, fasciculation, myelination, and axon guidance. 1 Mutations in the L1CAM gene cause L1 syndrome in humans. 2 This L1 syndrome includes widely heterogeneous phenotypes, such as HSAS (X-linked Hydrocephalus with stenosis of the aqueduct of Sylvius), Agenesis of Carpus Callosum, Spastic paraplegia, X-linked hydrocephalus, lower limb spasticity, mental retardation, and adducted thumb.3–5 To our knowledge, over 300 different L1CAM mutations have been published.6, 7 The majority of reported mutations are missense, nonsense, and small insertions or deletions. But the mutations in the extracellular part of L1CAM, which are expected to result in L1 protein truncation or absence of expression, are thought to cause a more severe phenotype in the L1 spectrum. The probability of detecting L1CAM mutations in isolated and clinically suspected cases was more than 74.2% 8 and is directly related to the number of L1 typical features of the patient and the patients with at least two additional cases in the family. 3 In the Indian scenario the study on L1 syndrome and L1CAM mutations is very limited, therefore it was imperative to take up the present study. We screened the different exons and exon-intron locations of the L1CAM gene for the mutations in Indian cohorts.
Methods
Patients
The present study was approved by the Institutional Ethical Committee (Ref no. KU/IEC/05-09/2014-15). Written informed consent was obtained from all the patients/ guardians involved. The data including age, sex, clinical history, and pedigree was recorded.
Karyotypic Analysis
To rule out the possible association of any chromosomal anomalies with L1 syndrome patients included in the present study, we tested the L1 syndrome patients by karyotypic analysis. 2.5 mL of blood was collected in a sodium heparinized tube (from BD Vacutainer, Kumar chemicals, Shivaogga, Karnataka) for lymphocyte culture and karyotyping. The microscope with image capturing technology and software ‘Ikaros’ by Meta systems was used to identify the chromosomal anomalies. Based on ISCN (An International System for Human Cytogenetic Nomenclature), 9 chromosomes were karyotyped and concluded as normal or syndromic.
Molecular Analysis
The blood sample of the patients was collected in a K3-EDTA tube (From Vacuette, Kumar Chemicals Shivamogga), and genomic DNA was extracted using a Qiagen QIAamp DNA Mini extraction kit (From Qiagen, Bengaluru). DNA was amplified by using a different set of primers (from SIGMA Aldrich, and Eurofins Genomics, Bengaluru) (Table 1). The primers were designed on the L1CAM GenBank sequence (Accession num: NM_000425.5) by using NCBI Primer-Blast (
Human L1CAM Gene Specific Primers Used in the Study.
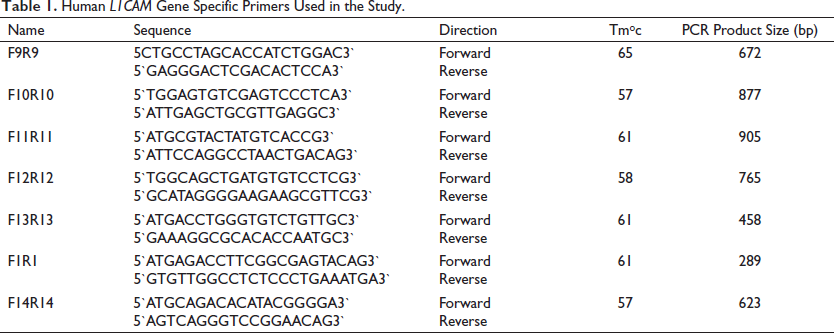
Cycle sequencing started with an initial denaturation at 96°C for 1 minute, followed by 25 cycles (denaturation at 96°C for 10 sec, annealing at 50°C for 5 sec, and extension at 60°C for 4 minutes) and followed by the hold step at 4°C.
Results
Study Population
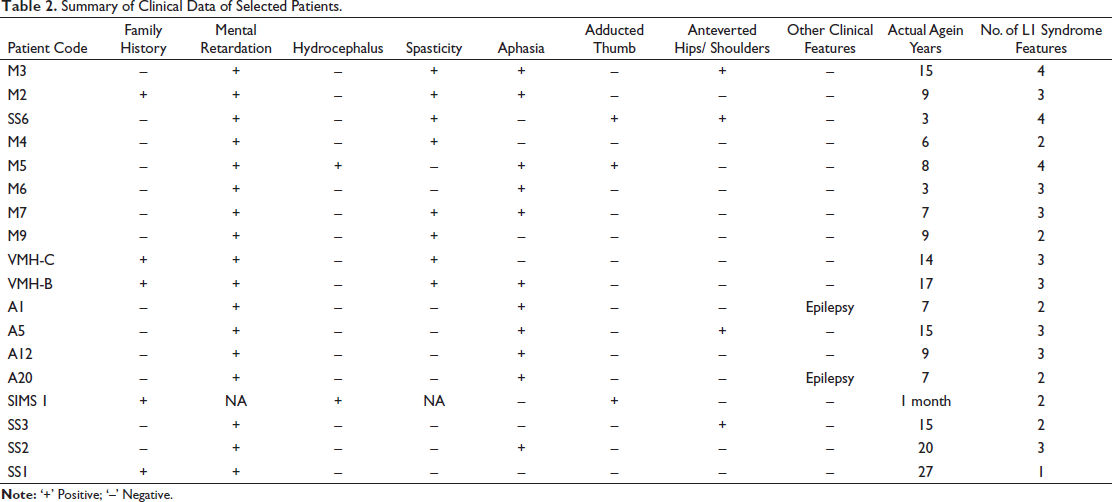
Based on L1 syndrome diagnostic criteria 113 patients were identified, and most of the patients shared one or more clinical features of L1 syndrome. Among them, 18 index cases (Table 2) with more typical features of L1 syndrome were screened for mutations in the L1CAM gene. The positive family history (71.42%) and the consanguinity (28.57%) may show an association in the development of the L1 syndrome (Table 3). In the present study, all patients displayed a combination of various L1 syndrome phenotypes. Among those, mental retardation is one of the primary and common clinical clues to identify L1 syndrome patients (Table 2). Mental retardation and aphasia (66.6%) are very prominent clinical symptoms observed in the present study.
Summary of Clinical Data of Selected Patients.
Patients with Consanguinity and Family History.
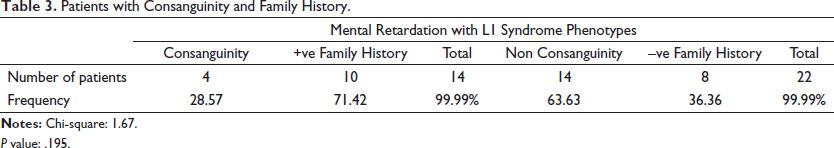
P value: .195.
Karyotyping
Karyotyping was performed to find out any chromosomal anomalies associated with the L1 syndrome and other diseases. The outcome of the study clearly revealed that all 18 analyzed patients had normal chromosomal arrangements (46, XY) and indicated that patients had no chromosomal anomalies and had prominent L1 syndrome phenotypes.
Screening of Mutations
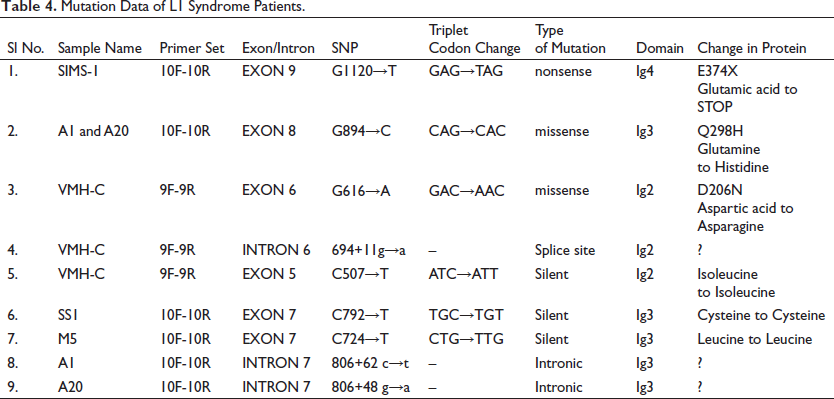
A total of 9 different L1CAM mutations were found in six index cases (Table 4). Six mutations were located in the coding regions, one mutation in the splice site region, and the remaining two were located in the intronic regions. Among nine mutations, two are missense, one nonsense, one splice site, two are intronic, and three are silent mutations.
Mutation Data of L1 Syndrome Patients.
Schematic Representation of Mutations and Protein Domain Model
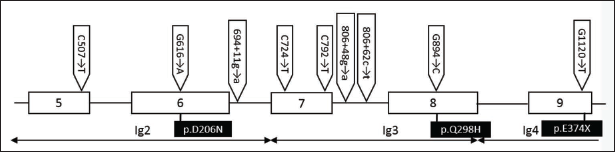
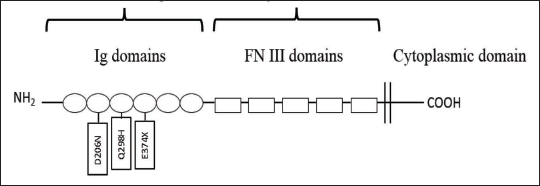
The mutations are depicted here according to the exon and intronic positions of the L1CAM gene (Figure 1). The rectangular boxes indicate the exons and the lines indicate the introns. Among all detected mutations, one nonsense and two missense mutations are protein-changing mutations and are found in the Ig domains of the L1CAM protein (Figure 2).
Protein Domain Model of the L1CAM Showing the Location of Missense and Nonsense Mutations.
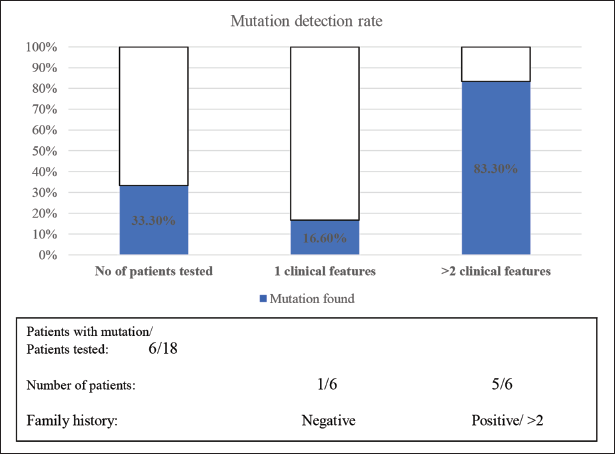
In the present study, mutations were identified in six patients among 18 patients studied (33.3%). Most of the mutations are private mutations that occur in only one family. The study revealed a high mutation detection rate (83.30%) in patients with more than two typical L1 syndrome features and more than two affected members in a family (Figure 3) compared to the least mutation detection rate (16.60%) in patients having a negative family history with only one typical feature.
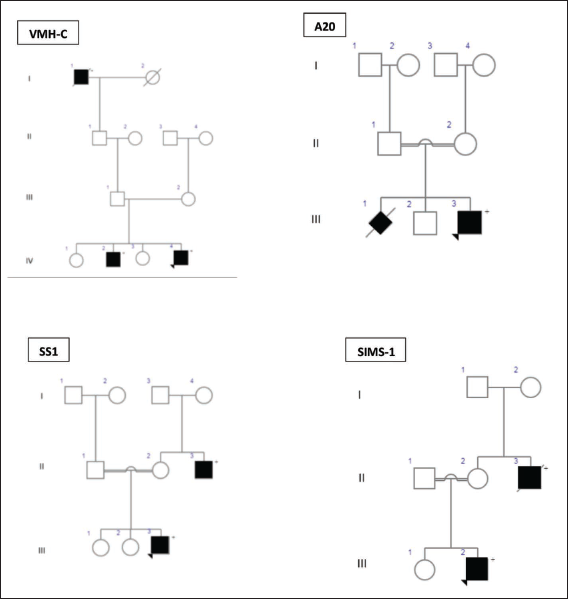
Illustration of Mutations
The mutation detection rate was related to the presence of typical clinical features of L1 syndrome. Mutations were found in 16.6% of patients with suspected L1 disease and a typical finding, in contrast to 83.33% of patients with more than two typical findings reported (X2 = 24.0, df = 10, P = 0.008) (Figure 4). Of these cases, the mutation detection rate was directly related to the number of L1 typical features reported.
Mutation Detection Rate Relative to Family History, and the Number of Reported Clinical Phenotypes.
In-silico Analysis
The present study identified one nonsense and two missense variants. The potential effects of these variants were investigated using different in-silico prediction programs. PROVEAN program predicted the variant (G1120→T) to be deleterious with a score of –3.192.
PolyPhen-2 program predicted the missense variant (G894→C) to be benign with a score of P = 0.004 (sensitivity 0.97 and specificity 0.59). And a score from the MUpro program was less than ‘0’ (–0.9484) indicating that the mutation decreases the protein stability. The confidence score was –0.6225 which is smaller and indicates a stronger prediction.
Another missense variant (G616→A) was found to be deleterious with a score below –2.5 by PROVEAN. The PolyPhen-2 predicted this mutation to be probably damaging with a score of 0.999. The MUpro also shows this missense mutation predicted to decrease the stability of the protein with a score of –0.9711 with a confidence score of –0.5686 which is less than ‘0’ and indicates more confidence in the prediction.
Discussion
The main features of the L1 syndrome include Corpus callosum agenesis, Mental retardation, Adducted thumbs, Spastic paraplegia, and Hydrocephalus. The clinical spectrum of L1 syndrome is highly variable and may involve any combination of these symptoms. At the severe end, there might be patients with massive hydrocephalus resulting in pre – or perinatal death, whereas, at the mild end, there are patients with mild mental retardation as their only abnormality. The phenotypes displayed in the L1 syndrome condition are overlapped with each other. Hence, the spectrum of clinical conditions varies in L1 syndrome.
In the present study, Sanger sequencing of the L1CAM gene in 18 patients allowed us to identify 9 different mutations reported for the first time and were never reported earlier (Table 4).
Two missense mutations (G894→C: transversion) and (G616→A: transition) were identified in the present study (Table 4). The transversion mutation G894→C was detected in exon 8 and is a novel missense mutation observed in two patients A1 & A20. Exon 8 encodes for the third immunoglobulin domain (Ig3) and this missense mutation has led to a change of Glutamine to Histidine in position 298 (Q298H). Since the Ig3 domain is responsible for homophilic interactions with other L1 proteins, this mutation probably affects the homophilic interaction of the domain.
Another mutation (G →A transition) was detected at position 616 (G616→A), of exon 6 of the L1CAM gene. Exon 6 encodes the second immunoglobulin domain (Ig2), and this missense mutation in exon 6 led to a change of aspartic acid to asparagine in position 206 (D206N). Aspartic acid is highly conserved in homologous adhesion molecules; therefore, the mutation probably affects the core structure of the domain. The Ig2 domain is responsible for homophilic interactions with other L1 proteins on the membranes of adjacent cells. Based on the mutation classification, both missense mutations (G894→C and G616→A) are novel mutations that come under class II extracellular missense mutations. To date, in the L1CAM mutation database, 81 missense mutations are reported.5, 8, 10–14 However, specifically on exon 8, only five missense mutations have been reported and on exon 6 only eight missense mutations have been reported (
Patient SIMS-1 carried a nonsense mutation (G1120→T) at the Ig4 domain in the extracellular portion at exon 9 which resulted in a stop codon yielding L1 protein truncated (E374X) (Table 4). Previous studies have found similar mutations in this region which manifest with variable clinical symptoms from Hydrocephalus to MASA syndrome.8, 10, 15, 16 The patient was diagnosed with hydrocephalus at the age of the first month, and at the age of 10 months, he was severely affected, both mentally and physically. And he belongs to a family with one affected male in generation II (Figure 3). Exon 9 encodes the 4th immunoglobulin domain (Ig4) and this nonsense mutation in exon 9 has led to a change of glutamic acid to STOP codon (GAG→TAG) in position 374 (E374X). Glutamic acid is involved in protein-active or binding sites and functions as an essential neurotransmitter and the body uses it to produce other neurotransmitters. This amino acid is also critical for brain development and function.
17
Therefore the interaction between L1 molecules has been disrupted due to truncated mutation (E374X) and resulted in abnormal development of the brain. This extracellular truncated mutation E374X represents a form of class I mutation.18, 19 (
The patient VMH-C carried a splice site mutation (694+11g→a) in Intron 6 of the L1CAM gene. This splice site mutation comes under class IV and influences the RNA splicing process. Class IV consists of extracellular mutations resulting in aberrant splicing of the pre-mRNA. These splice site mutations are in the highly conserved sequences flanking the intron-exon boundary, in the branch point signal.
27
The effect of these splice site mutations on the protein is unpredictable.28, 29 To date, in the L1CAM mutation database, 36 splice site mutations have been reported.4, 8, 10, 23 However, no previous study has reported any splice site mutation in intron 6 (
The present study has also found three silent mutations such as C507→T in the exon 5 of Ig2, C724→T, and C792→T in the exon 7 of Ig3. Since there is no successive change in the amino acid, these are silent mutations without any functional effects. To date, in the L1CAM mutation database, six silent mutations have been reported (
Two intronic mutations were found in the present study (806+48g→a; 806+62c→t) and belonged to the region Ig3. These intronic variations are present in the region of intron 7 and the effect of these mutations are unknown.
In the present study, mutations were found at the rate of one in six patients (16.6%) with L1 disease in contrast to eight mutations in five cases (83.33%) with more than two typical L1 features reported. Of these six patients, the mutation detection rate was directly related to the number of L1 features reported. The mutation detection rate in the present study is congruent with previous studies and confirms that the probability of finding L1CAM mutations increases by up to 66% in patients with three or more clinical features, and 74.2% in patients with at least two additional cases in the family.8, 10, 30, 31 Overall, the present study has reported the number of mutations that will enrich the L1CAM mutation database. These mutations will be a good source to investigate their functional aspects and their pathogenicity so that the mechanisms underlying the L1 syndrome can be understood further.
Limitations of the Study
The present study has limitations such as a small sample size and a limited number of exons of the L1CAM gene screened. However, this study offers a basis for expanding the study on L1 syndrome and identifying mutations further.
Conclusion
L1 syndrome has the most consistent features like hydrocephalus, mental retardation (MR), aphasia, spasticity, etc. Our findings support that L1 syndrome is associated with widely heterogeneous phenotypes. The present study has identified nine mutations, including one nonsense and two missense mutations. The present study is the first of its kind from the southern region of India to identify L1 syndrome patients and screen for mutations in the L1CAM gene. Our in-silico analysis revealed that some of the mutations detected are novel mutations. Since L1 syndrome exhibits extensive phenotypic heterogeneity, identifying L1 syndrome and mutations in the L1CAM gene would be necessary to understand the etiology of the disease. This would also be helpful in prenatal diagnosis and genetic counseling for families at high risk.
Abbreviations

Footnotes
Acknowledgements
We are thankful to the University Grants Commission (F. No. 41-95/2012 SR), New Delhi, India, for financial support.
Authors’ Contribution
Statement of Ethics
The questionnaire and methodology for this study were approved by the Institutional Human Ethics Committee Guidelines of the Kuvempu University and SIMS (Shivamogga Institute of Medical Sciences, Karnataka) (Approval number: KU/IEC/05-09/2014-15).
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The University Grants Commission (F. No. 41-95/2012 SR), New Delhi, India, provided the financial support.
ICMJE Statement
I/we had full access to all of the data in this study and I/we take complete responsibility for the integrity of the data and the accuracy of the data analysis.
Supplemental Material
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.