
Abstract
Background:
Progressive supranuclear palsy (PSP) is a rapidly progressive primary tauopathy characterized by vertical gaze palsy, postural instability, and mild dementia. PSP shows high clinical and pathologic heterogeneity. Although a few risk factors exist, such as advanced age and environmental toxins, the precise etiology remains largely elusive. Compelling evidence now suggests that genetic background plays a pivotal role in the pathogenetic pathways of PSP. Notably, PSP is genetically and phenotypically a complex disorder. Given the tau pathology, several studies in the past have identified microtubule-associated protein tau (MAPT) gene mutations/variations and its haplotype as the major genetic risk factor of PSP, both in the sporadic and the familial forms. Subsequently, genome-wide association studies (GWAS) also identified several novel risk variants. However, these genetic risk determinants fail to explain the pathogenetic basis of PSP and its phenotypic spectrum in majority of the cases. Some genetic variants are known to confer the risk, while others seem to act as modifier genes.
Summary:
Besides the complex genetic basis of PSP, the pathobiological mechanisms, differential diagnosis, and management of patients with PSP have further been complicated by genetic conditions that mimic the phenotypes of PSP. This is now becoming increasingly apparent that interactions between genetic and environmental factors significantly contribute to PSP development. Further, the effect of environmental factors seems to be mediated through epigenetic modifications.
Key message:
Herein, we provide a comprehensive overview of the genetic and epigenetic constructs of PSP and highlight the relevance of genetic and epigenetic findings in the pathobiology of PSP.
Introduction
Progressive supranuclear palsy (PSP) is a relatively rare neurodegenerative disorder characterized by abnormalities in movement such as vertical gaze palsy, poor balance, and frequent falls, vision, speech, and cognition. 1 Ocular abnormalities such as eyelid retraction, blepharospasm, apraxia of eyelid opening and closing have consistently been reported in patients with PSP.2, 3 Notably, these ocular abnormalities help distinguish PSP from closely related movement disorders like multiple system atrophy (MSA), Parkinson’s disease (PD), corticobasal degeneration, and dementia with Lewy bodies. 4 The prevalence of PSP has been reported to be 5.8 to 6.5 cases per 100,000, and the mortality rate is 3.1 to 6.5 per 100,000. 5 It is, after PD, the most common form of degenerative Parkinsonism and the third most prevalent form of neurodegenerative disease. 5 Neuronal loss in the basal ganglia and brain stem and wide distribution of neurofibrillary tangles (NFTs) in the brain areas such as subthalamic nucleus, striatum, thalamus, superior colliculi, globus pallidus, substantia nigra, oculomotor nuclei, periaqueductal grey matter, locus coeruleus, pontine nuclei, and raphe nuclei form the most common neuropathological hallmarks of PSP. 6 Similar to other neurodegenerative diseases, abnormal microtubule-associated protein tau (MAPT) has also been reported in patients with PSP. 7 Subsequent to this, another postmortem brain study demonstrated the presence of numerous intracellular NFTs and glial tangles comprising hyperphosphorylated tau protein. 8 Besides this, tau oligomers were identified in the brains of PSP patients. 9 Tau oligomers are found to be more toxic and can induce cognitive deficits in mice. 10 Notably, tufted astrocytes are considered reliable and specific markers for neuropathological diagnosis of PSP, and astrocytic tau pathology has been shown to correlate positively with NFT density as well as contribute to synapse loss in PSP.11, 12 It is important to note that these pathological changes and the accumulation of fibrillar aggregates of tau proteins in the brain contribute to different clinical phenotypes. 13 Clinical diagnosis of PSP often remains a challenge because of various phenotypes, and several clinical features are taken into consideration to recognize and differentiate it from related disorders like corticobasal syndrome (CBD), MSA, and dementia with Lewy bodies.
The precise cause of PSP is inadequately known in the majority of patients. The only predominant risk factor is advanced age. 14 The age of onset of PSP is generally between 45 and 75 years, with an average age of onset of about 63 years. 15 Although it has traditionally and consistently been considered a sporadic disorder, an increasing number of reports indicate its familial clustering. 16 The familial aggregation of PSP provides preliminary evidence toward the involvement of genetic factors in its pathobiology and inheritance. 17 Subsequently, findings from genetic studies demonstrated important implications of genetic factors not only in the risk and progression of PSP but also in cognitive dysfunction in the patients. 18 Amongst the earlier genetic findings, mutations in MAPT gene were considered the strongest risk determinants of PSP. 19 However, with the advent of high-throughput genomic technologies, several other genetic loci were linked to PSP, which includes but is not limited to myelin-associated oligodendrocyte basic protein (MOBP), syntaxin 6 (STX6), and eukaryotic translation initiation factor 2-α kinase 3 (EIF2AK3) genes. 20 Despite these advances, there exists a high degree of pathological, clinical, and genetic heterogeneities. A recent study suggests the involvement of diverse molecular mechanisms, including transcriptional dysregulation and epigenetic modifications, in driving distinct cell-specific tau pathology in PSP. 20 The involvement of epigenetic processes implies the role of environmental factor(s) in PSP pathobiology. Association between various environmental factors and the risk of PSP was reported by many studies.21, 22 It seems likely that the genes and environmental factors might confer risk to PSP either alone or through their potential interactions. However, there is a considerable lack of data on the precise role of these components in the pathophysiology of PSP. Herein, we attempt to highlight the role of genetic, epigenetic, and environmental factors and dissect their specific contribution to the clinical and pathological heterogeneity of PSP.
Methods
Search Strategy
A systematic literature search was performed from January 1964 to July 2020 using PubMed, Embase, and Cochrane Library databases following the preferred reporting items for systematic reviews and meta-analysis (PRISMA) guidelines (Figure 1). Search terms, such as PSP, mutation, genome-wide association studies (GWAS), polymorphism, epigenetics, gene expression, miRNA, PSP look-alikes, and environmental factors were used. Important and relevant studies missed by the database search were screened manually from the reference lists of the retrieved articles. The prospective references were selected by assessing titles, followed by abstracts and full text based on the eligibility criteria described further.
PRISMA Flow Diagram Showing Screening and Selection of the Articles for the Study.
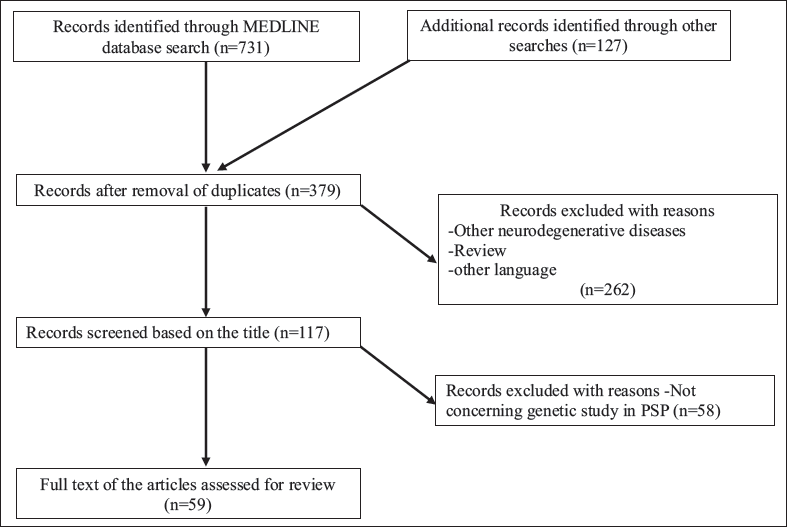
Eligibility Criteria
Following inclusion criteria were used for this review: (a) search was limited to human, experimental animal, and in vitro studies, and (b) papers published in the English language. The selected list of papers was used for exploring additional references.
Genetic Risk Factors of PSP
Genetic studies adopting classical strategies such as twin and adoption are rare in PSP. There is no definite heritability estimate for PSP. Contextually, heritability has been found to vary between different pathological subtypes of frontotemporal tauopathies. Over the past few years, there have been several attempts to understand the genetic underpinning of PSP by candidate gene, linkage as well as GWAS. The pathogenic mutations having direct causal effects as well as the common and rare variants [single nucleotide polymorphisms (SNPs) and copy number variations (CNVs)] modifying the risk of PSP have been identified. Thus, the data obtained from the genetic studies suggest a very complex genetic basis of PSP. It is important to note that these findings do not support its monogenic nature, and based on the current data, the genetic definition of PSP may require revision. A detailed and updated appraisal of the genetic risk determinants of PSP has been highlighted in the following sections.
Findings from Candidate Gene Association Studies
Role of the MAPT Gene in PSP
PSP is a primary tauopathy, where the prominent pathological feature is the presence of fibrillar aggregates of the MAPT. The MAPT gene coding for tau protein is a long gene (134 kb) and has 16 exons on chromosome 17q21. Mutations in the MAPT gene were found to cause frontotemporal dementia with Parkinsonism linked to chromosome 17 (FTDP-17); this indicated that MAPT mutations leading to tauopathy are involved in neurodegeneration. 23 The hominid tau protein is characterized by an N-terminal region, a proline-rich domain, a microtubule-binding domain (MBD), and a C-terminal projection region. In the adult human brain, a single MAPT gene generates six tau isoforms by alternative splicing of exons 2, 3, and 10. The isoforms differ based on the exclusion or inclusion of a repeat region (either three or four imperfect repeat regions). In a normal adult brain, the ratio of 3R and 4R are approximately equal (1:1); however, their ratios are altered in most tauopathies.24, 25 In both in vivo and in vitro studies, a higher 4R:3R tau ratio contributed to PSP pathology because of the inclusion of exon 2 and exon 10 of the MAPT gene.26, 27 Notably, the altered ratio increases the potency of tau phosphorylation and aggregation, decreasing the binding affinity of tau to the microtubule (MT). Destabilized tau-MT structure hampers the normal MT function, leading to neurodegeneration.
Polymorphisms and Haplotypes of the MAPT Gene in PSP
Following the identification of the MAPT dinucleotide polymorphisms in intron 9 as a risk determinant of PSP in 1997, 19 several other polymorphisms covering the entire coding region of the MAPT gene were reported. Two polymorphic inversions in the MAPT gene have resulted in two major haplotypes, H1 and H2, which differ in orientation, and recombination does not happen between these haplotypes. 28 Notably, H1 tau haplotype is predominantly associated with PSP and CBD.29, 30 Both the H1 and H2 haplotypes have several subhaplotypes, and some of these have functional implications in the context of pathology. For example, the frequency of the H1c subhaplotype is less than 10% and associated with an elevated risk of PSP. 29 In a study on Spanish PSP patients, H1E’A subhaplotype was found to be present in 16% of PSP patients, while H2E’A was rare in the patients, implying that the latter might play a protective role. 31 A very recent study suggested novel associations between three H1 subhaplotypes, such as H1d, H1g, and H1o with PSP. This study further indicated that several MAPT haplotypes could potentially influence the severity of tau pathology. 32
In addition to the above, several other SNPs conferring risk to PSP have been reported within the MAPT gene. Many SNPs are in complete linkage disequilibrium (LD) and tag the H1 and H2 haplotypes. In a study, rs2240756 and rs110402 were found in low LD with del-In9 but showed a strong association with the disease variants in H1 haplotype. 33 Some studies have also used haplotype tagging SNPs (htSNPs) in understanding the genetic risk of PSP. A study adopting haplotype tagging approach examined six htSNPs; this included five htSNPs representing intra-H1 variation (rs1467967, rs242557, rs3785883, rs2471738, and rs7521) and a 238-bp insertion/deletion polymorphism within intron 9 (del-In9), and observed that two common haplotypes from these htSNPs were significantly associated with PSP. 29 The above studies suggest that these polymorphic variants might contribute to increased risk, or alternatively, the polymorphic risk variants are in LD with a causal variant of PSP.
Besides SNPs, the CNVs, because of loss or gain of genome segments (from 1 kb to several kbs), have a significant impact on the genetic makeup of an individual as well as on the phenotypes. Few studies have shown robust associations of CNVs with various neurological disorders.34, 35 However, data pertaining to CNVs in PSP are albeit limited. In a recent study, the MAPT duplication was identified in PSP, indicating that CNVs can also serve as potential risk determinants of PSP. 36
Mutation Spectrum in the MAPT Gene and Risk of PSP
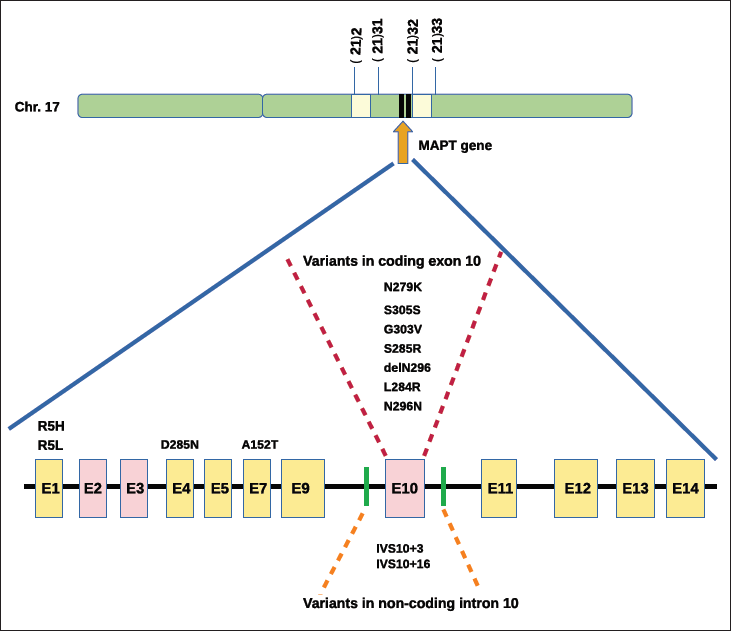
To date, 111 mutations/variations have been identified in the MAPT gene, of which more than 50 mutations have been reported to be pathogenic (Alzforum, mutation database,
Shows Genomic Location of the MAPT Gene and 13 Reported Mutations Within the MAPT Gene in PSP.
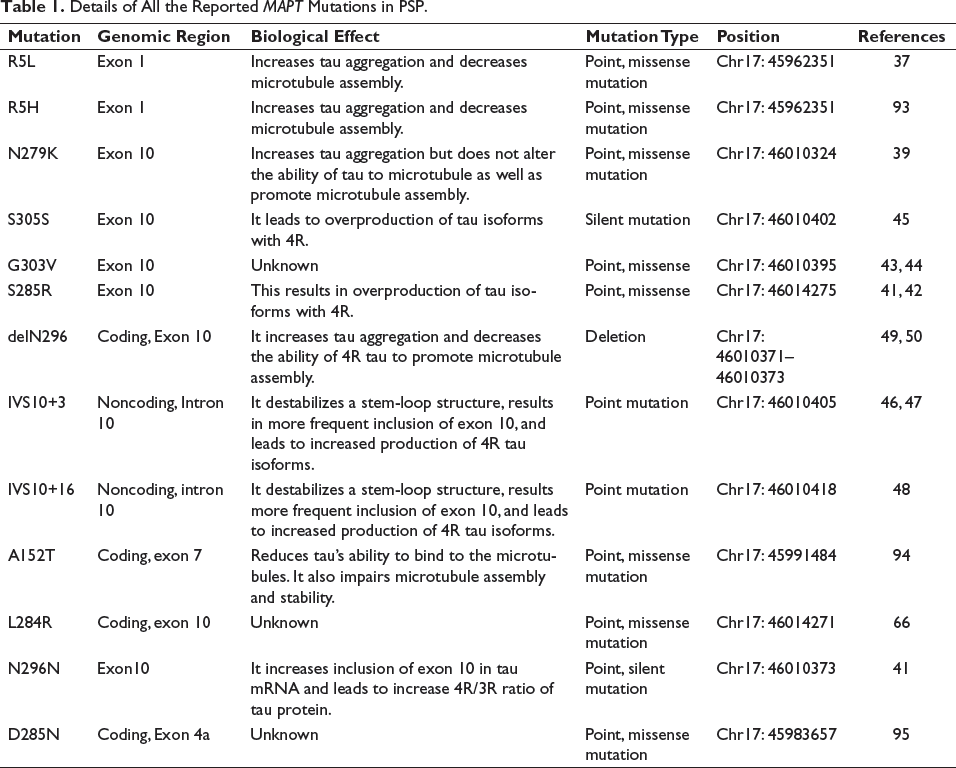
Details of All the Reported MAPT Mutations in PSP.
Missense Mutations
p.R5L Tau Mutation
R5L (g.45962351G>T, c.14G>T), a mutation in the amino-terminal end resulting in an alteration of tau interaction with tubulin, and tau MT, was detected in a Caucasian female patient with PSP. 37 This mutation lowers the MT formation but does not affect 4R:3R ratio. The isolated tau from patients carrying this mutation was mostly of straight and irregular length. In a study, R5L mutation was shown to directly accelerate the nucleation process of tau aggregation. 38
p.N279K Tau Mutation
N279K is an asparagine-to-lysine mutation at position 279, within exon 10 of the MAPT gene. N279K (46010324T > G, c.1842T > G) mutation was first found in a sibling of a French family, having cognitive as well as memory dysfunction and vertical supranuclear palsy. 39 N279K mutation leads to overproduction of the tau isoform with four MT-binding motifs. However, this mutation neither alters the binding ability of tau to tubulin nor promotes MT assembly. In vitro studies have demonstrated that neuronal stem cells with N279K, derived from iPSCs, had displayed an increased ratio of 4R:3R in the cells, including morphological changes such as cellular stress, accumulation of stress granules, and vesicle trafficking deficits. 40
S285R Tau Mutation
The first S285R mutation was found in a 46-year-old Japanese man who had no family history of dementia or movement disorders but with probable PSP. 41 In another study on three sib-pairs with autopsy-confirmed PSP, S285R was reported in a sib-pair whose mean age at symptom onset was 41 years. 42 Pathologic examination revealed severe neuronal loss and gliosis in globus pallidus and subthalamic nucleus. Biologically, this mutation was found to affect exon 10 splicing and overproduction of tau isoforms containing 4R tau. 41
p.G303V Tau Mutation
Two studies have reported a novel mutation, G303V, located in exon 10 of the MAPT gene. The first study was on a family with three pathologically confirmed PSP that identified G303V mutation of tau (46010395G>T,c.821G>T), and the proband carrying this mutation was found to have increased expression of 4R tau protein. 43 The second study was on another family having six clinically heterogeneous PSP patients over three generations. 44 G303V mutation, resulting in a transversion of two amino acids in exon 2, has been shown to control tau expression and phosphorylation. Notably, G303V mutation was associated with early-onset PSP in one of the studies.
Silent Mutations
p.S305S Tau Mutation
S305S (g.46010402T > C, c.828T > C) is a novel silent mutation located in the last codon of exon 10 within the stem-loop of the MAPT gene. This was first described in a pathology confirmed patient with dystonia in one hand, falls, dysarthria, and supranuclear gaze palsy, having a positive family history. 45 Although S305S mutation does not result in an amino acid change in the tau protein, the presence of this mutation was shown to cause 4.8-fold increased splicing of exon 10 by functional exon trapping experiments, leading to an increased 4R:3R in familial PSP patients.
Intronic Mutations
IVS10+3 Tau Mutation
IVS10+3 (g.46010405G > A, c.1920 + 3G > A) is a splice site mutation located in the intron following exon 10 on the MAPT gene. This intronic G-to-A transition dislocates the stable stem-loop structure, resulting in excessive inclusion of exon 10, followed by overexpression of 4R tau isoform. 46 This mutation was first identified in a multiple-system tauopathy with presenile dementia (MSTD) family. 46 Subsequent to this, Spina et al. 47 examined MSTD family, comprising more than 200 individuals and spread over seven generations, and observed that of the 21 affected individuals, 17 individuals had frontotemporal dementia, and 2 individuals had the symptoms of an atypical form of PSP. In contrast, 2 others had either a severe postural imbalance or an isolated short-term memory deficit. 47
IVS10+16 Tau Mutation
IVS10+16 (g.46010418C > T, c.915 + 16C > T) is one of the most common intronic variations found in familial cases of PSP all over the world. In the disease condition, this point mutation hinders the stem-loop structure involved in regulating alternative splicing of exon 10 and results in enhanced inclusion of exon 10 and 4R:3R ratio in the brain. 48
Deletion Mutations
delN296 Tau Mutation
delN296 (g.46010371_46010373ATA, c.1889_1891ATA) results in the deletion of three nucleotides, leading to a protein sequence devoid of asparagine at position 296 in the second tubulin-binding repeat of tau protein. It was first found in a 38-year-old patient born from a third-degree consanguineous marriage and had a PSP-positive sibling. 49 This was further reported in 2 members of a family having the same condition as above, including repeated falls. 50 This mutation is supposed to be pathogenic in homozygous condition, whereas in the heterozygous form, it has only a mild penetrance to PD-like condition. This mutation caused a 2.5-fold overexpression of 4R tau compared to the wildtype, resulting in abnormal interaction with MTs. 51
Findings from GWAS and Other Genetic Studies
The genetic risk of PSP was predominantly linked to the MAPT gene and its haplotypes. However, some studies have failed to identify pathogenic mutations in the MAPT gene in sporadic PSP. 52 This indicates that besides the spectrum of pathogenic mutations within the MAPT gene, additional genetic variations seem to be involved in influencing the risk of PSP. To address this knowledge gap, genome-wide linkage as well as GWAS studies were carried out. In most of the GWAS studies, multiple SNPs were reported to be present more frequently in PSP; this suggests that polymorphic variants might also modify the risk of PSP.
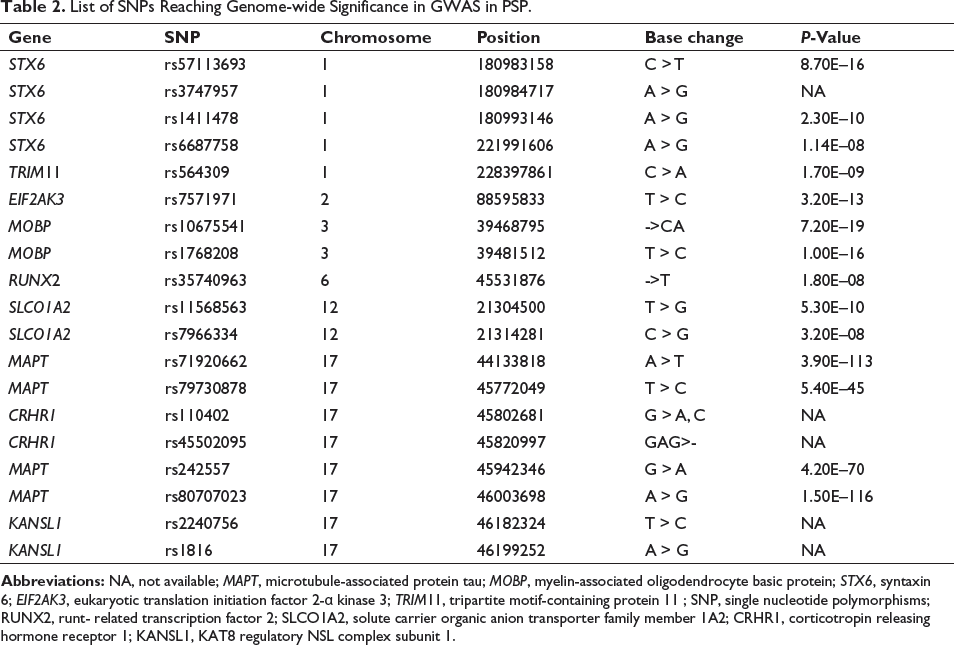
The first GWAS was carried out in 2007 in the American and Canadian populations, 53 and MAPT H1 haplotype was strongly correlated with the disease phenotype. In addition, a second main locus on chromosome 11p12-p11 also exhibited high signal of association at allelic (P < .001), genotypic (P < .001), and haplotypic (P < .001) levels. Another GWAS identified three more risk loci, such as STX6, EIF2AK3, and myelin-associated oligodendrocyte basic protein (MOBP), besides the MAPT gene in European populations. Interestingly, two SNPs, rs242557 (PJ = 4.2 × 10–70) and rs80707023 (PJ = 1.5 × 10−116) located within the MAPT gene, were found to affect the risk of PSP. Of these SNPs, rs242557 is a regulatory polymorphism that influences the brain expression of MAPT. 20 On the other hand, a recent study found rs8070723 to be significantly associated with older age of onset. 54 STX6 is located in chromosome 1q25.3 and codes for a soluble N-ethylmaleimide-sensitive factor activating protein receptor (SNARE) class protein, STX6. It is envisaged that genetic variations within STX6 may affect protein trafficking machinery, including the movement of misfolded proteins, leading to endoplasmic reticulum (ER) stress.55, 56 EIF2AK3 gene encodes protein kinase R (PKR) like endoplasmic reticulum kinase (PERK)-a component of the ER unfolded protein response (UPR). PERK gets activated when there is excess accumulation of unfolded proteins; this inhibits protein synthesis and allows the ER to clear misfolded proteins. However, the precise mechanism(s) through which genetic variations within EIF2AK3 confer(s) risk to PSP is unknown. Another gene found to be associated with PSP risk is MOBP; this encodes a protein, produced by oligodendrocytes. The expression of MOBP has been reported to be high in the brain regions, such as the white matter of the medulla, pons, cerebellum, and midbrain that are affected in PSP. 57 Variation in MOBP gene length contributes to PSP pathogenesis by altering myelin or oligodendrocyte function.
A recent joint GWAS identified five gene loci that reached genome-wide significance threshold P < 5 × 10–8.20, 58 Of these five risk loci, three loci (MAPT, STX6, and MOBP) were identified in a previous GWAS, 20 and the remaining two are novel loci, runt- related transcription factor 2 (RUNX2) at chr.6p21.1 and solute carrier organic anion transporter family member 1A2 (SLCO1A2) at chr. 12p12.1. Few SNPs such as rs79730878 (P = 5.4 × 10−45) and rs71920662 (P = 3.9 × 10−113) located in extended haplotype containing the MAPT region, rs10675541 (P = 7.2 × 10−19) within MOBP, rs57113693 (P = 8.7 × 10−16) within STX6, rs35740963 (P = 1.8 × 10−8) within RUNX2, and rs7966334 (P = 3.2 × 10−8) within SLCO1A2 reached genome-wide significance (P < 5 × 10−8). 58 Subsequently, another replication study also reported the association of PSP with MAPT, MOBP, STX6, and EIF2AK3 genes. 59 This study also identified two novel risk variants, rs11568563, located within the SLCO1A2 gene, and rs687758, located within the DUSP10 gene. Subsequently, a recent GWAS has reported an intronic variant (rs564309) within the tripartite motif-containing protein 11 (TRIM11) gene in chr. 1q42.13 that strongly influences the clinical phenotype in PSP. 60 TRIM11 is a component of the ubiquitin-proteasome system (UPS), a critical mediator of tau pathology, and has predominant expression in the cerebellum and basal ganglia. It is suggested that common variation within 1q42.13 locus seems to modify the function of TRIM11 differentially in specific brain regions, and the variants within this locus act as a genetic modifier of the clinical phenotypes of PSP.
In addition to GWAS, genetic studies on candidate genes were also conducted on patients with PSP, and these studies have led to the identification of some novel variants. A recent study comprising a discovery cohort (N = 246) with possible and probable PSP patients as well as a validation cohort (N = 704) with definite PSP patients examined the role of LDL receptor-related protein 10 (LRP10) gene on PSP patients. In this study, two possibly pathogenic variants, p.Gly326Asp and p.Asp389Asp in two patients of the discovery cohort and seven possibly pathogenic variants, such as p.Arg158His, p.Cys220Tyr, p.Thr278Ala, p.Gly306Asp, p.Glu486Asp, p.Arg554*, and p.Arg661Cys in eight patients from the validation cohort were identified. 61 Mutations in the leucine-rich repeat kinase 2 (LRRK2) gene have consistently been found to cause both familial and sporadic cases of PD. Impact of LRRK2 gene variations on the risk of PSP was explored in a few studies. Although a novel p.A1413T mutation within the LRRK2 gene was reported in a PSP patient, 62 other studies failed to report any association of LRRK2 gene variations with PSP risk and suggested that LRRK2 gene may not contribute to PSP risk.63, 64 In addition to these genes, missense variants of ABI3 and PLCG2 genes, previously associated with Alzheimer’s disease, were shown to be associated with patients diagnosed neuropathologically with Lewy body disease (LBD-NP) but not with PSP. 65 In a Caucasian family from the South of England, multiple family members across different generations had PSP syndrome, and one member was shown to have a novel L284R mutation in exon 10 of the MAPT gene. This mutation was suggested to be associated with the young age of onset and prominent behavioral features. 66 A list of SNPs reaching genome-wide significance in GWAS of PSP has been summarized in Table 2.
List of SNPs Reaching Genome-wide Significance in GWAS in PSP.
Role of Gene-Environmental Interactions in PSP
The environmental risk hypothesis of PSP has also gained momentum in recent times. Multiple studies have demonstrated an increased risk of sporadic PSP because of exposure to environmental toxins, including drinking well water for long years, lesser educational attainment, consumption of traditional medicines, etc.21, 67 The consumption of plants containing isoquinoline derivatives, toxic for dopaminergic neurons, and acetogenins that act as inhibitors of mitochondrial respiratory chain has led to overrepresentation of atypical Parkinsonism of PSP-type. 68 It is also being envisaged that sporadic PSP could also be mediated partially by genetic as well as environmental risk factors. Further support towards this was garnered from animal model studies demonstrating an association between increased risk of PSP and exposure to plant-derived neurotoxic substances such as annonacin, a mitochondrial toxin that induced tauopathy in a rat model. 69 In a recent study in Wattrelos in Northern France with severe environmental contamination by industrial metals, a cluster of 92 sporadic patients with PSP was reported. 70 The authors reported an overrepresentation of the H1 haplotype and H1c subhaplotype found in the chromosomes of 95.8% of cases. From the above studies, it is not clear whether genetic mutations make an individual more susceptible to PSP in the presence of environmental toxins, including heavy metals. To resolve this knowledge gap, an in vitro study has examined the neurotoxic effects of chromium, nickel, and cadmium on two different cell models, such as SH-SY5Y neuroblastoma cells, and induced pluripotent stem cell (iPSC)-derived neurons (iNeurons) carrying MAPT mutation. These three heavy metals induced cell death in iPSC-derived iNeurons, and iNeurons with R406W MAPT/tau mutation were found to be more prone to cell death. Exposure to heavy metals also led to apoptotic cell death in SH-SY5Y cells. Additionally, exposure to Cr and Ni led to increased tau protein expression and phosphorylation in both the iNeurons and SH-SY5Y cells. 22 These data suggest an important contribution of environmental toxins, including heavy metals in the risk of PSP.
Role of Epigenetic Processes in PSP
Although the expanding role of genes and environmental factors helped to understand the multifactorial basis of PSP, the precise mechanisms through which these factors mediate the risk of PSP are yet to be discerned. Environmental factors alter the expression of genes by influencing epigenetic processes, predominantly DNA methylation. A study analyzing targeted DNA methylation of MAPT gene in the brain tissue demonstrated differential methylation of the MAPT gene in PSP patients. 71 A specific CpG island, called CpG1, located within intron 0 of the MAPT gene has shown significant hypomethylation in frontal cortices but not in the occipital cortices in the brain tissues of patients with PSP. Interestingly, this hypomethylation correlated with increased mRNA expression of the MAPT gene in PSP-affected brain tissues. Besides, a genome-wide methylation study identified differentially methylated probes (DMPs) clustered within the chr.17q21.31 region in the peripheral blood of patients with PSP. 72 This finding is very interesting because this chromosomal region contains MAPT, the major risk gene of PSP. Subsequently, another epigenome-wide methylation study on postmortem forebrain tissues of PSP patients demonstrated significant differential methylation at 717 CpG sites, of which 451 are linked to protein-coding genes. 73 These 451 sites correspond to 375 annotated genes; of these, significantly pronounced differential methylation was found at DLX1 gene, which differed by up to 10% between PSP patients and controls. It is noteworthy that DLX1 has two specific binding sites in the promoter region of MAPT, and in an in vitro study, DLX1 was shown to negatively affect MAPT expression.
GWAS has identified several SNPs as putative risk variants of PSP. However, the mechanism through which these SNPs modulate the risk to PSP is not known. A study was conducted to understand whether PSP-linked SNPs correlate with brain gene expression and CpG methylation. 74 Methylation levels of a CpG in the 3´region of ARL17B exhibited a significant association with rs242557 and rs8070723 SNPs. Methylation levels of this CpG for rs8070723 were lower in carriers of H2 haplotype, while the same CpG had higher methylation levels in rs242557-A carriers. In addition to this, methylation levels of an intronic CpG with rs1768208, as well as another intronic ARL17A CpG associated with rs242557, were also reported.
Besides methylation, gene expressions are controlled by differentially expressed micro-RNAs (miRNAs). A recent study revealed upregulated expressions of miR-147a, and miR-518e, while downregulated expression of miR-132 in forebrain tissues of PSP patients. It was further demonstrated that the expressions of the target genes of miR-147a (NF1, ACLY, and ALG12) and the target genes of miR-518e (CPED1, JAZF1, and RAP1B) were repressed in the forebrains of PSP patients. 75 In addition, miR-132, directly involved in the regulation of the level of neuronal splicing factor polypyrimidine-tract-binding protein 2, was found to be upregulated in PSP. 76 Furthermore, variable methylation of mitochondrial tRNAs (mt-tRNAs) at 11 site in the ninth position (“p9 site”), which affects the efficiency of the translation as well as downstream mitochondrial function, was observed in PSP patients. 77
“PSP-Look-Alikes”: Genetic Conundrum
The clinical picture of PSP is relatively well-defined, and a definite diagnosis is made based on neuropathological features. However, detailed clinicopathological findings have led to identifying various “atypical” clinical manifestations of PSP. 78 Such conditions are referred to as “PSP-look-alikes.” Many genetic conditions carrying mutations in MAPT, PGRN (progranulin), chromosome 9 open reading frame 72 (C9ORF72), ATP13A, and DCTN1 (dynactin) genes may present with a PSP phenotype. 79 Few examples of PSP-look-alike disorders are highlighted below.
Frontotemporal Lobar Degeneration
These are caused by mutations in PGRN, C9ORF72, CHMP2B, or FUS genes. Of these, C9ORF72 hexanucleotide (GGGGCC) expansion is well characterized in frontotemporal dementia. The pathophysiology because of changes in this gene develops when the repeats of expansion are >60 compared to 2 to 23 repeats in the controls. A recent investigation suggested a direct association of this mutation, having an extension of more than 50 repeats with PSP patients. 80
Perry Syndrome
Perry syndrome also represents a PSP-like phenotype. A number of studies have suggested that variations within the dynactin subunit 1 (DCTN1) gene can lead to Perry syndrome.81, 82 It is noteworthy that a variant, p.K56R of the DCTN1 gene was also identified in PSP patients. 83
Kufor-Rakeb Syndrome
Kufor-Rakeb syndrome (KRS) is a rare, juvenile-onset, and levodopa-responsive type of Parkinsonism. It is characterized by pyramidal degeneration, dementia, supranuclear gaze palsy, and cognitive impairment. 84 It is caused by autosomal recessive mutations in the ATP13A2 gene. Two loss-of-function mutations (c.1306 + 5G > A in exon 13 and 3057delC/1019GfsX1021 in exon 26) as well as a 22-bp duplication in exon 16 (1632_ 1653dup22 or 55LfX788) 85 were reported in patients with KRS.
Niemann-Pick Type C (NPC) Disease
Niemann-Pick type C (NPC) disease is another example of a PSP-look-alike condition. It is also a rare autosomal recessive neurodegenerative lysosomal storage disorder associated with supranuclear vertical gaze palsy. 86 In majority of the cases with NPC, mutations in the NPC1 and NPC2 genes have been reported.87, 88
Gaucher Disease
Gaucher disease is a lysosomal storage disorder that follows autosomal recessive inheritance. Individuals with adult-onset Gaucher disease can be characterized by prominent cognitive dysfunction and slowness of vertical saccades, mimicking PSP. Gaucher disease is caused by mutations in glucosylceramidase beta (GBA) gene. 89
Creutzfeldt-Jakob Disease (CJD)
Supranuclear gaze palsies have also been reported rarely in familial or sporadic Creutzfeldt-Jakob disease (CJD). 90 Previous studies have demonstrated associations of mutations at codons 129 and 200 in the PRNP (prion protein) gene on chr.20 with progressive supranuclear vertical gaze palsy in patients with familial CJD.91, 92
Conclusion
Even after 50 years of its discovery, the pathobiology, diagnosis, and management of PSP remain enigmatic. It has now been realized that combined effects of genetic, epigenetic, and environmental factors are involved in the susceptibility and pathogenetic pathways of PSP. These advances have undoubtedly helped in resolving diagnostic uncertainty to a large extent. There is a growing perception that genetic testing on a definite panel of genes differentiating PSP and PSP-look-alikes would make a significant stride in their diagnostic and prognostic management. Further genetic and epigenetic studies are required, which would potentially improve the diagnosis of PSP and also lead to the development of targeted therapies.
There is a dramatic increase in genetic data in tauopathies; some genetic variants are shown to have pathogenic relevance, while others are involved in influencing the disease risk. The list of polymorphic variants, including both the SNPs and the CNVs conferring the risk, is gradually expanding in neurodegenerative diseases. It is being increasingly realized that analysis of polygenic risk score seems essential in interpreting the genetic risk of neurodegenerative diseases. Given the quantum of risk variants identified by GWAS, future studies should focus on performing polygenic risk score analysis in PSP. Besides this, an attempt should be made to adopt a PSP-specific genetic algorithm for stratifying monogenic PSP from genetically complex PSP cases.
Footnotes
Authors’ Contribution
MD, SD, and RY had conceived the idea for the article; MD and SD performed the literature search and data analysis, MD and SD drafted and/or critically revised the work. MD, SD, NS, PKP, and RY critically revised the work.
Statement of Ethics
Not applicable
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work is supported by the Indian Council of Medical Research (grant number:5/4-5/197/Neuro/2019-NCD-1).