
Abstract
The approval mechanism for new drugs in India has in recent years been a subject of scrutiny and controversy in both the public sphere and academics. Frequent rollbacks in the guidelines have attracted criticisms from the industry as well. All this happens at a time, when the country aspires to catch up and keep pace with the fast-changing global regulatory norms. This article draws upon the scholarships on regulatory science and institutional theories to understand the regulatory processes of new drugs approval in India. Both strands of scholarships highlight the multidisciplinary, context-specific character of regulatory knowledge. We find that Indian regulation-making process relies heavily on regulatory knowledge built up elsewhere, potentially limiting the scope of generating its own, context-specific, regulatory knowledge through learning and capacity building. Inadequate level of deliberations in the decision-making process, dearth of documentation and a lack of connections with the larger institutional settings appear to be two other features of it. All this might explain the reasons for frequent changes, and lack of continuity, in regulation for new drugs approval in the country in recent years.
Introduction
While new drugs popularly mean new molecular entities, in reality, it takes various forms, ranging from reformulated generics to fixed-dose combinations (FDCs) of existing drugs. This article focuses predominantly on the regulatory processes of approving new generics and FDCs for two reasons. 1 First, these drugs constitute the overwhelming majority of new drugs launched in the country till date. Second, the approval process for new molecular entities merits a separate discussion. There have been major controversies regarding the regulatory processes of approving new generic drugs and FDCs. Academic literature, however, has remained relatively indifferent to study these processes.
The emerging literature on institutional economics and ‘regulatory science’ 2 highlights the institutional character of regulation-making process. Institutions are stable pattern of thoughts and actions, based on shared beliefs or legal constraints, established to reduce uncertainty in behaviour (Hodgson, 1988, 2010; North, 1990). Social scientists’ interest in technical standards and regulation has increased in the recent years, and there is a consensus that advancement in science and technologies are not the sole and perhaps, not even the main, drivers of science-based regulations (Daemmrich, 2004). Regulations become authoritative when empirically verified facts are filtered through social processes of informal negotiations and shaped into frameworks arising out of shared opinions and compromised outcomes (Jasanoff, 1987). These shared opinions and compromised outcomes are the results of settling differences in opinion and inferences within a socio-economic and political sphere (Chavance, 2008).
Scientific claims themselves can be constructed socially and may represent ‘political capture’. 3 Regulation making for science-based industries has been studied widely in the context of developed countries. However, in the case of developing countries, social science literature on regulation-making processes has only started emerging in the recent years. This is not surprising given that regulation making has in fact, started only recently in many developing countries. The present article aims to contribute to this growing scholarship by analysing the new drug approval processes in India.
In India, the drug approval processes have been marked by several controversies involving frequent rollbacks of, and back-and-forth movements in, guidelines and recommendations (Thatte & Bavdekar, 2008; CDSCO, 2012; Desai, 2012; Joshi, 2012; Kadam & Karandikar, 2012; Mattheij, Pollock, & Brhlikova, 2012; Shenoi, 2012). The literature on institutions, on the other hand, emphatically notes that an institution derives its credibility by offering stable pattern of behaviour. This study is an attempt to analyse the regulation-making process for approval of new drugs in India through the prism of institutional economics and science and technology studies on regulation making to explain, and draw relevant policy lessons, from such events.
This article has six sections. The second section outlines the conceptual framework of the study. The third section has two subsections. In subsection ‘New Drugs in India: The Meaning and Nuances’, we briefly delineate the regulatory concept and definition of new drugs, both in India and in some of the countries with major pharmaceutical market. Subsection ‘The Institutional Character of New Drug Approval: A Brief Overview’ briefly discusses the major regulatory processes in approval of new drugs and describes how the institutional underpinning of their process leads to its evolution. The fourth section outlines the sources of data and the method of our analysis. The fifth section is the key section, which analyses the new drug approval processes in India. Finally, the sixth section draws the broad conclusion.
The Conceptual Framework
Institutions and regulations are socially devised mechanisms that structure human interaction, and promote durable and routinised pattern of behaviour (Hodgson, 1988, p. 10; North, 1990), and habits, often through ‘downward causation’ of rules, norms and behavioural pattern set by the ‘authority’ (Hodgson, 2010). They are made up of formal laws and informal norms and conventions and their enforcement characteristics. Prevailing social beliefs, knowledge, power, interest and attitude towards a particular phenomenon all shapes an institution and determines its evolution.
The process of regulation making is referred to as ‘regulatory science’. Scientific exercises contribute to the knowledge of policymaking. Produced in complex institutional settings, they follow different validation criteria than those followed by basic or research sciences. It dwells in the interspace of industry, government, civil society and the academia. While academic science is characterised as open-ended, innovative, subject to peer review and undertaken with the aim of advancing knowledge—the regulatory science on the other hand, is characterised as bounded by external pressure of time and politics, directed towards closure, proprietary, subjected to a variety of types of review and undertaken with the aim of aiding, and making laws and regulations based on contemporary policies. Regulatory science is therefore socially negotiated. Its production of knowledge is not confined to conventional sites like scientific industries or educational institutions. Wider implications of scientific claims are debated to analyse its social relevance, acceptability and robustness of the knowledge so prescribed. The society brings in its own narrative. For Jasanoff (1990, p. 19), regulatory science is ‘a hybrid activity that combines elements of scientific evidence and reasoning with large doses of social and political judgment’. An important feature of regulatory science is the heavy involvement of government and industry in the production, validation and consolidation of knowledge. Regulatory authorities are believed to be accountable, and, therefore, influenced by various non-scientific actors: parliament, media, judiciary and the civil society, often as part of their constitutional obligation (refer Chowdhury, 2016). The discourse for validating science in regulatory context is therefore fluid, debatable and politically encumbered. It has a direct bearing on the economic activity through making, and remaking of, institutions.
The definition of institution is often an exercise marred by controversies and opposing ideas, and in that sense, there is hardly any definition of institution that attracts consensus. However, some of the common characteristics of institutions emphasise that they are the ‘rules of the game’. Institutions are formal or informal rules that constrain (or augment) economic behaviour to reduce uncertainty in economic activity. From a process perspective, institutions shape the cumulative process of adaptation of means to ends, or an ensemble of knowledge, set of habits and rules of morals, custom and law, which have a common centre or goal, or an outgrowth of habits. Custom and norms are considered to be essential to economic life, and institutions have their customary practices and laws. They are the results of adaptation processes, being in turn responsible for the selection of social structure and individual economic activity. Thus, from a process perspective, co-evolution and reciprocal determination coexist in the realm of institutions. Both the formal legal rules and the informal social norms, while shaping each other, govern individual behaviour and structure social interactions (Hodgson, 2006; North, 1990; Veblen, 1898).
Preferences, along with expectations of the future, habits and motivations, not only determine the nature of institutions but also are limited and shaped by them. If people live and work in institutions on a regular basis, it shapes their world views. Institutions arise, develop and function in a pattern of social self-organisation beyond conscious intentions of the individuals involved. Vacillations of institutions are necessarily a result of the very incentives created by such institutions and are thus endogenous (Chavance, 2008).
For the present study, we deal with the Indian rules and guidelines concerning bioequivalence (BE) for drugs already registered in the Indian market and the approval processes for FDCs. 4
New Drugs and Their Regulatory Approval: An Overview of Their Institutional Character
New Drugs in India: The Meaning and Nuances
In principle, the definition of new drugs in India is similar to its definition in other countries. We briefly discuss the definition and characteristics of new drugs in India.
Rule 122E (Drugs & Cosmetics Rules) defines the ‘new drug’ to mean and include:
drug, including bulk drugs substance (or phytopharmaceutical drug) which has not been used in the country to any significant extent under the conditions prescribed, recommended or suggested in its labeling and has not been recognised as effective and safe by the licensing authority for the proposed claims, drugs already approved for certain claims, which are now proposed to be marketed with modified or new claims (viz., new indications, dosage, dosage forms or routes of administration), or fixed dose combinations of two or more drugs, individually approved earlier for certain claims, which are now proposed to be combined for the first time in a fixed ratio, or a new fixed ratio, with certain claims, (viz., indications, dosage, dosage forms or routes of administration).
5
It is further noted that ‘a new drug shall continue to be considered as new drug for a period of four years from the date of its first approval or its inclusion in the Indian Pharmacopoeia whichever is earlier. 6 ’
It is important to note that the term ‘generic drug’ has not been defined or mentioned in Indian regulations. Rule 122-A prescribes that the first applicants seeking a new drug’s registration in India have to submit data including the results of local clinical trials. According to Appendix I-A of the Schedule Y (data required from applicants of a new drug already approved in the country), subsequent applicants seeking registration of the same drug do not require to submit results of local clinical trials, but only need to provide data from bioavailability (BA)/BE and comparative dissolution studies for oral dosage forms.
‘Investigational new drug’ is described under an explanation at the end of Rule 122DA, which pertains to application for permission to conduct clinical trials for New Drug/Investigational New Drug. The explanation states for the purpose of these rules, Investigational New Drug means a new chemical entity or a product having therapeutic indication but which have never been earlier tested on human beings.
Finally, FDCs are a category of ‘new drugs’ that combine two or more active pharmaceutical ingredients (API). Hailed by a section of the industry for its ability to enhance patient compliance and reduce medical costs, these drugs evoke mixed response among, physicians, regulatory authority and civil society, across the globe. While their prescription is common even among the most qualified physicians for facilitating the patient compliance or to manage some adverse events, some social activists and a section of the industry have always been sceptical about them. In India, their approval has been a controversial process. Allegedly, many Indian states approve such drugs without following the due procedure. In addition, one observes conflicting views and positions even between the central drug regulatory body and the states’ regulatory bodies.
The Institutional Character of New Drug Approval: A Brief Overview
Conventionally, drug regulation was almost synonymous with national sovereignty (Vogel, 1998). From the mid-1990s, however, a new trend began with heightened cooperation between the regulatory agencies of the developed countries, culminating in the formation of the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH). For many scholars, this harmonisation has enhanced regulatory efficiency of many countries, leading to faster approval of drugs, without greater risks. However, these countries also had different histories of regulation-making processes, with varying protocols, validation methods and importance given to different entities involved in the regulatory process (Daemmrich, 2004; Ray & Bhaduri, 2003), much of which seem to continue in the post-harmonisation era (Ratanwijitrasin & Wondemagegnehu, 2002). A huge diversity in national regulatory capacity does exist, and not all national regulators can effectively implement even minimum regulatory oversight of pharmaceutical market in their jurisdiction. Some scholars argue that harmonisation among the advanced countries has often prioritised trade interests’ safety. Abraham and Reed (2001), for instance, argue that harmonisation has lowered the standards of drug safety in Europe and made public health goals secondary to trade interests. Science, according to them, has become subservient to politics in deciding the standards of safety in the harmonised framework. To elaborate their points, Abraham and Reed quote a representative of the European Federation of Pharmaceutical Industry Associations (EFPIA):
It isn’t pure science. There you are in the US where drugs have always been tested with a year’s toxicity and suddenly because of some negotiating with Europe, you’re now reducing the safety margin on drugs being tested. … I think the EU had to be very careful about the public reaction which says ‘hey wait a minute, all these years we’ve had drugs on the market which were only tested for 6 months and now you’re telling us they should have been tested for 9 months’—you know—and that’s why the EU absolutely have not said that drugs have to be tested for 9 months—they will accept 6 months. They will continue to accept 6 months. (as cited in Abraham & Reed, 2001, p. 121)
The authors point out several areas where the ICH settled for less stringent mechanisms, where a more stringent mechanism has been in practice in some of the member countries, purportedly, to facilitate faster approval process, allegedly, in the interest of the industry. Rago and Santoso (2008) in this context caution that excessively high number of new drugs may distort health care practices, and raise health care costs unnecessarily. 7
Increasing globalisation of pharmaceutical production, however, has continued unabated, and with it the attempt to globalise the standards of ICH. The WHO (2002) in this context highlights several issues where the implementation of ICH standards in developing countries can pose serious regulatory challenges and can raise serious public health concerns. These concerns emanate from high resource and knowledge intensity of the regulatory structure of ICH to, at times, lack of adequate data to validate the claims of benefit of following ICH procedure, including the requirement of involving various ethnic groups in clinical trials (pp. 12–21). The need for country-specific regulation remains a relevant point of discussion in the debates on regulation making.
In India, new drugs launched elsewhere are subjected to clinical trials alone and on local patients (typical of Phase 3 trials). The requirement for local clinical trials is based on the claims that ethnicity and environmental factors can influence efficacy and safety of drugs. In actual practice, however, this criterion has not always been strictly adhered to. The Parliamentary Standing Committee on Health and Family Welfare in its fifty-ninth report of May 2012 noted
according to information provided by the Ministry, a total of 31 new drugs were approved in the period January 2008 to October 2010 without conducting clinical trials on Indian patients. Two drugs (Ademetionine and FDC of Pregabalin with other ingredients) were somehow not included in the list. Thus there is no scientific evidence to show that these 33 drugs are really effective and safe for Indian patients.
The report also noted ‘A review of the opinions submitted by the experts on various drugs show that an overwhelming majority are recommendations based on personal perception without giving any hard scientific evidence or data.’ We, therefore, observe negotiation among science, parliamentary committees, regulatory bodies as well as personal preferences and perceptions of regulatory officials in making, and following, the regulatory guidelines on new drugs even in India. No study, however, has attempted to analyse the process in detail.
Similar negotiations have been observed in the making of regulatory guidelines for BE studies. The concept of BE has evolved in a sociopolitical and economic sphere wherein several approaches to the analysis and treatment of data were considered by the FDA. Some major approaches included the power approach, the 75/75 rule, the confidence interval approach and the Bayesian approach. Since then, tremendous advancements have been made by the FDA, and currently, the approaches to determine BE of pharmaceutical products have been largely standardised.
The BE refers to the absence of a significant difference (/-20%) in the rate and extent to which the active ingredient (of the test drug vis-a-vis the reference drug) becomes available at the site of drug action when administered at the same dose. 8 Underlying the concept of BE is the claim that if a drug product contains a drug substance that is chemically identical and is delivered to the site of action at the same rate and extent as another drug product, then it is equivalent and can be substituted for that drug product. Some studies in the USA demonstrated that the presence of different non-active ingredients in a medicine as well as the process of manufacture can alter the dissolution rate of the active ingredients, which initiated the search for a biological test assay to validate generic medicines in the USA, and finally led to the creation of the concept of BE in the 1970s (Carpenter & Tobbell, 2011). 9 It was incorporated in the regulatory framework of new drug approval in the USA in the year 1984.
Emergence of the concept of BE for approval of generic drugs dates back to the Hatch Waxman Act in the USA, which for the first time introduced differentiated requirements for generics and the new chemical entities. Generics, in the changed scenario, required only undertaking BE tests, while a new chemical entity was poised to undergo a much costlier and lengthier process of clinical trials. The equivalence of the rate and pattern of absorption of a new drug, with that of the ‘original’ drug, into the body seeks to prove its therapeutic equivalence with the original drug. In more than one sense, passing of this Act heralded a new beginning for the generic industry in the USA. The Act also ‘sought to encourage generic manufacturers to challenge pioneer patent rights by conferring a six month period of market exclusivity on the first generic manufacturer to challenge the pioneer patent rights …’ (Danzis, 2003, p. 588).
The spread of the regulatory framework on requirement of BE for registration of generics in the USA has, however, not been smooth. Carpenter and Tobbell (2011) narrate how science, interests and public health concerns all negotiated with each other to give this law a shape. However, impact of the concept of BE on access to drugs continues. Over time, USFDA has come up with the criteria to waive the requirement of BE (bio-waivers) for some drugs to prevent delay in their access. Later, the concept of bio-waivers has been applied to some essential medicines in the list of WHO, once again, to ensure that the cost of conducting BE does not stand in the way of their easier access by the poor. Although the criteria for giving ‘bio-waivers’ include scientific parameters like solubility and permeability of a drug, the WHO and USFDA guidelines differ in their ranges. 10 This, once again, hints at the possibility of socio-economic and institutional factors shaping science-based standards in the field of new drugs approval.
The transfer of BE requirements to the developing countries is a relatively recent phenomenon. Among the middle-income countries, Brazil took a long time to adopt this law in a full-fledged manner, reportedly, to provide its domestic industry with the time and scope to develop necessary technological capacity to conduct BE studies. Although, of late, Brazil has developed adequate domestic capability to conduct BE studies, its costs are relatively high. The new regulation in Brazil requires BE to be conducted in Brazil for all drugs produced in the country.
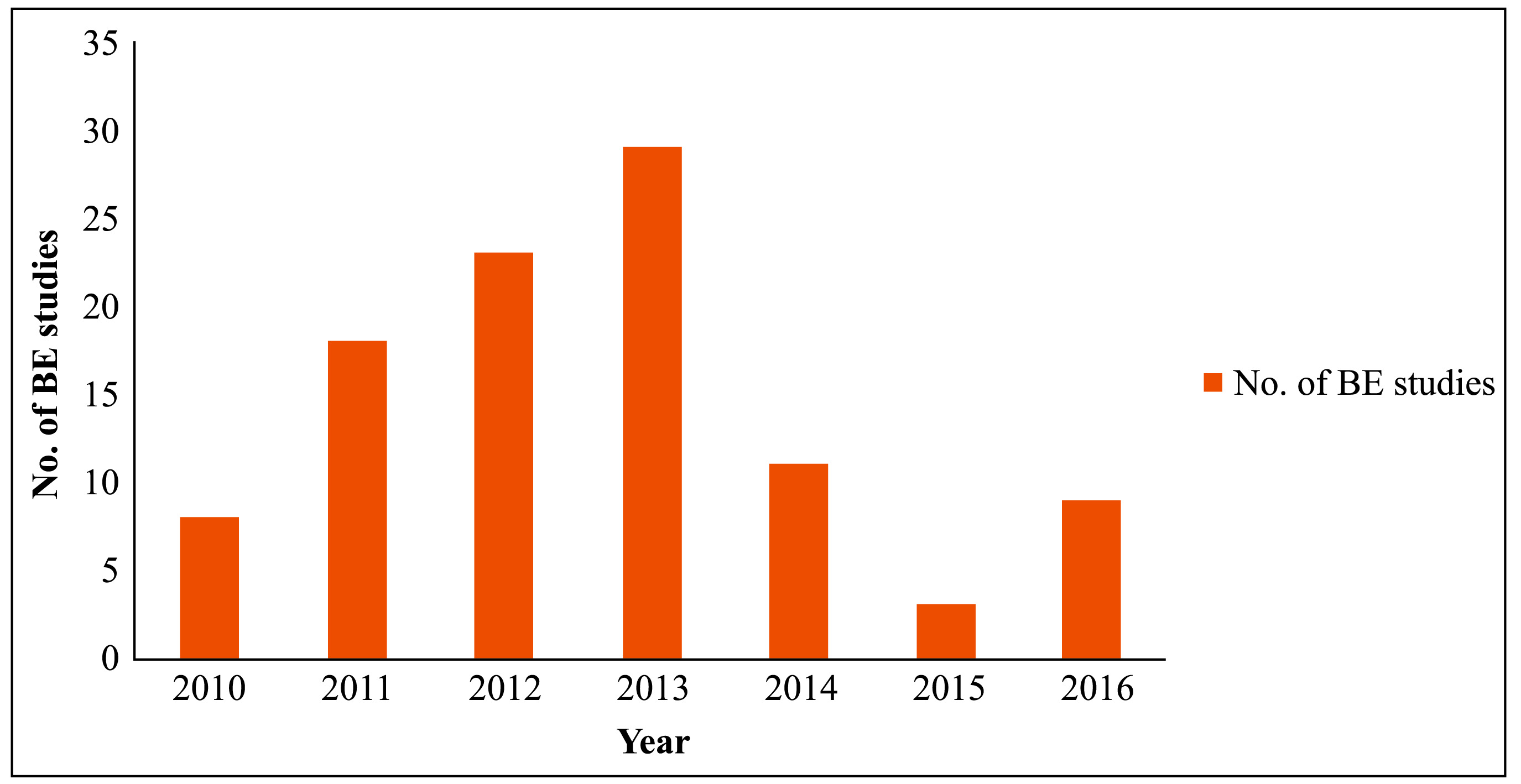
Indian clinical research industry seized on the opportunity to conduct BE studies, and several organisations set-up laboratories, purportedly, to become a global hub for BE studies. Even though the Indian regulations require BE data only from the applicants who apply during the first 4 years following a new drug’ registration, these laboratories expected business from exports through drug registrations in foreign countries. Clinical research regulations in India, which also cover BE studies, have recently seen quite a few back-and-forth movements. Many in the industry and media often allege that this institutional instability has hampered the growth of the ‘BE industry’. Approvals granted for conducting BE studies in the last 5 years are depicted in Figure 1.
Data and Methods
Data for this study come from primary as well as secondary sources. For secondary data, we relied mostly on the website of the Central Drugs Standard Organization (CDSCO)—an organ of the Directorate General of Health Services under the Ministry of Health and Family Welfare, Government of India. For primary data, we visited various firms, interviewed independent experts and heads of Clinical Research Organisations (CROs) in various parts of India on a project funded by the ANRS, France. We interviewed the heads of five CROs and two hospitals each from Kolkata, Delhi and Mumbai. In addition, we interviewed four independent experts in the field, and one clinical investigator.
Discussion and Analyses
Regulation Making for Bioequivalence
The BE data are required to be established only by those subsequent applicants who seek approvals within the first 4 years of the launch of a new drug in India. 11 No BE study is required for generics, whose ‘reference formulations’ have been in the Indian market for more than 4 years. The underlying principles and procedures for generating BE data for Indian regulatory submissions are quite in line with the global practices.
Our reading of the guidelines of the CDSCO suggests further that drugs are categorised into three groups in terms of their launch in India
12
:
Drugs not yet approved in India, but approved elsewhere. Drugs approved in India within 4 years of the first approval of the same molecule. Drugs approved in India after 4 years of the first approval of the same molecule.
For the first category of drugs, an applicant has to submit ‘Clinical study data and published report of pharmacokinetic and pharmacodynamic study carried out in healthy volunteers, and data published in reputed journals’ to the authority. 13 For drugs belonging to the second category, only BE data is required. The drugs in the third category can enter the market with only a proof of chemical equivalence. It appears that clinical practice of these drugs in the Indian market are taken to be a substitute for the requirement of new clinical data.
All scientific experts we have interviewed more or less agree that establishing BE is essential, and without this, the quality of drug product (formulation) would remain untested if we only bank on chemical equivalence. Despite this assertion, however, we find existence of covert concerns with the requirement of BE data.
A university scientist, who has set-up a dedicated clinical research unit for BE studies, expressed scepticism about the use of currently adopted BE standards to study drug efficacy, especially on poor Indian patients, who have not had previous exposure to modern allopathic medicines. According to him, because of previous non-exposure, compliance to the conventional BE standards may imply overmedication to this section of people. But he said he could not substantiate his claims nor could he publish his findings because most of the contract research he does is sponsored with publication restrictions. His view evokes mixed responses. While some leading pharmacists agree with his findings some others find it irrelevant. The head of one leading CRO supports this view in a roundabout manner. Regarding the objections on conducting BE studies in Indian population for drugs being exported or BE study services being exported, he cautioned that such trials might involve additional risks if people in the destination country require higher dosage. He, however, cites no concrete reason for such differences. This issue needs serious investigation from public health researchers. 14
India until now has not been requiring BE data for drugs, which are launched 4 years after the first launch of the reference drugs or their inclusion in the Indian Pharmacopoeia (IP), whichever is earlier. 15 This is popularly known as ‘the 4-year clause’.
The reasons for this clause, however, do not seem to have been made available in any public repository of documents. Noted pharmacists, subject experts, industry officials and regulators we spoke to were not aware of any concrete reason, and drubbed it as ‘unscientific’, ‘anomalous’ and ‘bureaucratic compulsion’. Although several attempts have been made in the recent past to amend this provision, it remained unchanged ‘in want of wider consultation’ (Chaudhury, 2013). Even these policy documents do not cite any reason for the emergence or continuity of this clause. 16 Our interaction with an industry expert, who was involved with regulation making in the 1990s as a WHO consultant, finally, provided an insight. He recalled his conversations with two retired Central Drug Controllers and argued that these exemptions in the 1980s were made to safeguard public health. The central unit (CDSCO) has the authority to regulate imports, exports and ‘new drugs’ while the state units issue manufacturing licences. The central unit has always had more resources and regulatory capacity than its state counterparts. It is also alleged that irregularities and procedural lapses have been more common on the part of the state units. As a consequence, the central regulatory body, in its wisdom, apparently had apprehended that the state units in those days did not have enough regulatory or technical capacity to regulate new drugs. Indeed, pharmacopoeia was not regularly updated in those days. The manufacturing protocol for new drugs, therefore, was not easily available in the public domain. It was also thought that competition between the various state regulatory units might lead to undesirable haste in approving new drugs. Under such a scenario, this rule was believed to a prudent way to restrain the state units from approving drugs. The rule, according to this view, helped to protect public health from trade interest and improper regulatory practices. This 4-year period, according to them, gave the Central Drugs Laboratory 17 enough time to establish and validate the testing protocols for new drug substances for firms to follow. 18 Indeed, in the recent past, inclusion of new drugs in IP has had widely varying gap years after their first approval in India. Table 1 captures the time it has taken for some of the drugs to be included in IP after their first registration in India.
Lag between First Registration of Select Drugs and Their Inclusion in Indian Pharmacopoeia
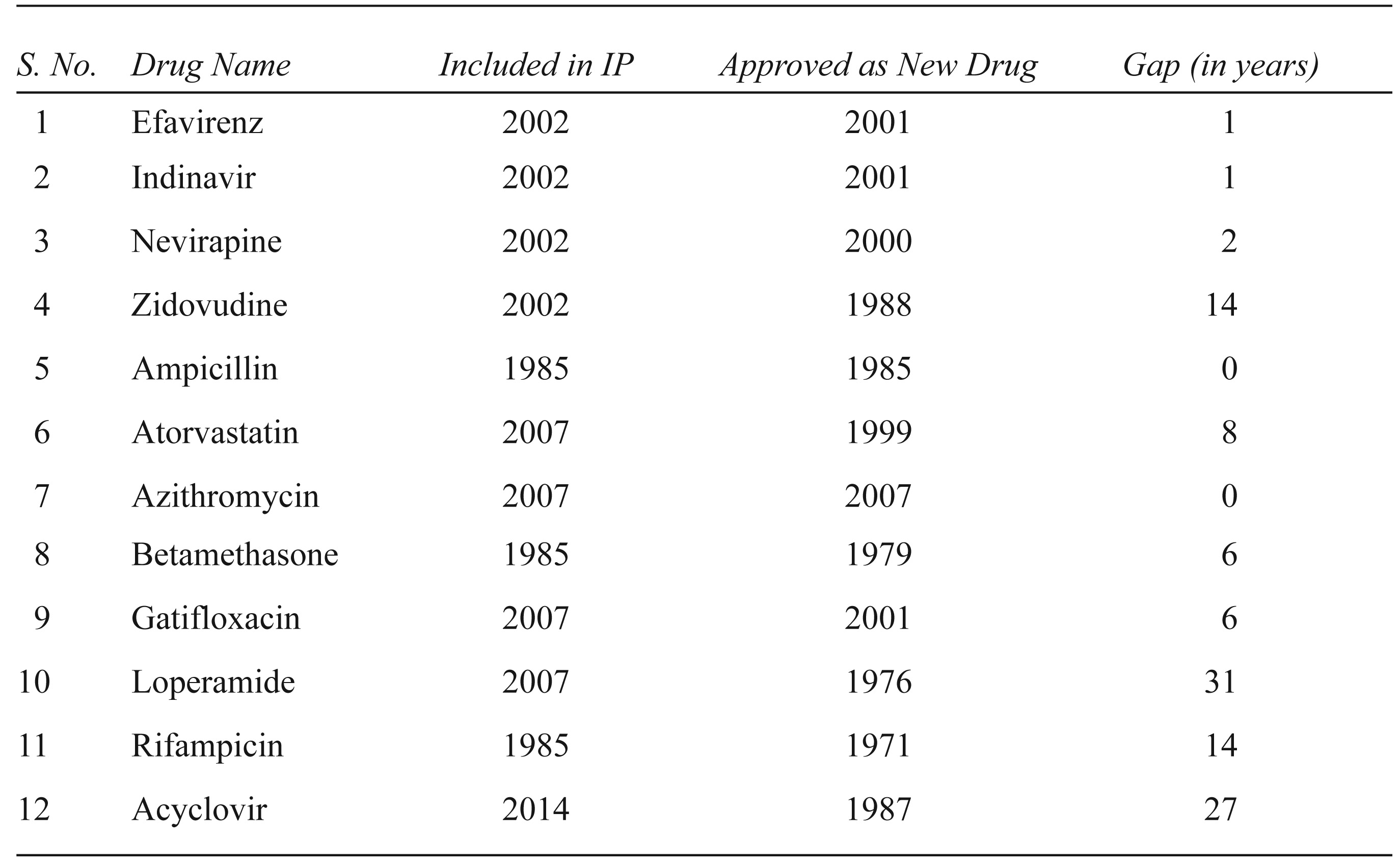
It is important to note that the process of inclusion of methods to prepare new drugs into the pharmacopoeia has significantly improved over the past 10 years after constitution of the Indian Pharmacopoeia Commission. With more periodic establishment and standardisation of drug specifications, the reference to the time limit until a new drug is included in IP may have become redundant.
The continuation of the ‘4-year clause’ may, therefore, provide an example of an outlived institution. Its inclusion in the first place was also perhaps an attempt, a veiled one though, to admit a lack of control of the central regulatory authorities over its regional branches, and perhaps mistrust in the latter’s regulatory mechanism: by defining all drugs launched within the 4 years of the first launch as new drugs, the central regulatory authority made it mandatory for drug manufacturers to seek approval through the central authority. While one may find a strong public health interest among the regulation makers in trying to stop misuse of regulatory powers of the state authorities, the rule may also be argued to have jeopardised subsequent efforts to strengthen the regulatory structure of the regional bodies by weakening their role in the approval process. Moreover, the non-documentation of regulatory steps or non-availability of the documents for past deliberations reveals that everything is not ‘institutionalised’ enough in the arena of pharmaceutical regulation in India.
On bio-waivers, Indian regulation has so far adopted a more stringent view similar to the United States Food and Drug Administration (USFDA) but is now seeking to include the dimensions highlighted by the European agency, the WHO and the Japanese regulatory authority and thereby relax its guidelines to broaden the ambit of bio-waivers in order to bring down the cost of generics even further (Chaudhury, 2013, p. 38). In addition, India seems to provide a passive form of bio-waivers to all drugs that are ‘not new’ after 4 years of their first registration. However, the ministry, in want of wider consultation, has not yet accepted these recommendations (Chaudhury, 2013, pp. 11–12).
Approval Mechanism for Fixed-dose Combination Drugs in India
The FDCs are a contentious issue in pharmaceutical practice and regulation. On the one hand, FDCs are hailed for improving patient compliance in certain disease like tuberculosis and HIV-AIDS, and reducing health care costs, on the other hand, FDCs have attracted criticism from health activists, law makers and some sections of the industry on grounds of being ‘irrational’ and ‘dangerous’ largely due to undesirable drug–drug interaction. In reality, it occupies a significant part of the market for pharmaceuticals in India. The share of FDCs in Indian pharma market can be gauged by the fact that the recent government decision to impose a ban on 350 odd FDCs by the Indian regulatory authority has resulted in succumbing the growth of the Indian pharmaceutical sector to its 2-year low. 19 The ban has, however, been revoked recently by the Court.
First applicants for each FDCs must conduct a full, four-phase clinical trial if the FDC is not marketed in India, and one or more of its active ingredients are not approved in any country. However, only the Phase III clinical trial (popularly called ‘bridging studies’) is needed if the FDC is marketed in other countries. 20 For FDCs whose active ingredients are approved and marketed in India, the regulation asks only for in vitro studies if the FDC is marketed abroad. In case, it is not marketed in any other country but has a history of concomitant use in the country; only ‘adequate evidence’ of their safe and effective concomitant use may be necessary for their approval, if published data is not available. For seeking approval of minor changes for the FDCs marketed in India, only in-vitro studies are required. The same guideline applies to FDCs which are combined only to bring convenience, and whose individual ingredients have been in concomitant clinical use.
According to the guidelines issued by Indian regulatory authorities, a BA/BE study is also required when ‘some changes are sought in an FDC, which is already in the Indian market’, especially if the justifications for the claims made are ‘not satisfactory’.
What it reveals is an interesting interaction between science, clinical practice as well as ‘regulatory wisdom’ to shape the process. Long-term clinical practice of concomitant use of the drugs used in an FDC can make a case for its exemption from clinical trials, and BE studies and the same holds if an FDC has been approved and marketed in other countries. A deeper investigation reveals the complex relationship that the central regulatory authority has had with its state-level counterparts. In many cases, it is contended that the regulatory authorities in the states have provided licences for FDCs without exchanging relevant information with the central authority. The exceeding of authority by state regulatory officials together with inadequate regulatory capacity of state regulatory authorities has, allegedly, aggravated the situation, where many ‘irrational’ combinations have been approved without the approval from the central regulatory body which is mandatory for all new drugs. However, the ground for terming them irrational has evoked mixed response. A section of the industry and even the Court have questioned the attempt to label these combinations irrational without presenting any data for their irrationality. In fact, their long-term clinical use, without much concrete evidence of ‘safety hazards’, has made judicial interventions a difficult task. Unavailability of well-articulated scientific evidence may make it difficult for the judiciary to pronounce any conclusive judgement, leaving the impasse to continue. 21 The other component of irrationality is ‘ineffective product’. Many reports in the media and by the health activists have alleged that combination drugs are ineffective. Once again, however, rarely have these claims been substantiated by credible data. Regulators have in such a situation, depended on physicians’ feedback. The CDSCO, admittedly, has accepted such reports to continue with several FDCs, for example, Aceclofenac and Drotaverine combination, and the Rajya Sabha Report on CDSCO mentions that positive feedback was received from many physicians of various government hospitals. However, the Report has also questioned the modalities of organising those feedbacks as well as their neutrality and impartiality of providing the feedbacks. In other words, the Report hints at a possible nexus between a section of the officials of the regulatory authority, industry and some physicians. Some studies have also brought out the lack of awareness among the medical professionals encouraging continued use of certain FDCs without caring about their rationality (Dalal, Ganguly, & Gor, 2016). The physicians, according to this study, seem to rely only on medical representatives to form their opinions about drug efficacy and rationality. While lack of awareness and the issue of nexus between industry and physicians is well known and has been a subject of intense research across the globe, the basis of the Rajya Sabha Report’s doubts on the impartiality of this feedback is not based on any data. Rather, taking a legalistic stance, the Rajya Sabha Report draws its conclusion relying on procedural loopholes of collective feedback, and, somewhat more importantly, the fact that the particular combination has not received approval in countries with more established regulatory systems in the global North. This aspect of relying more on the practice of regulatory systems in other countries than attempting to generate own clinical data reveals regulatory dependence and can forestall the possibility of regulatory learning and emergence of new scientific knowledge. 22 Indeed, through our search for academic literature on effectiveness of this combination, we find a paper by Pareek et al. (2010) establishing therapeutic effectiveness of the aforementioned FDC to treat primary dysmenorrhea. Using other countries as reference, therefore, may impinge the progress of regulatory knowledge. It is, perhaps, in order here to mention that the dependence of Indian regulatory authorities on data generated by other countries is a common phenomenon, across the industry. 23
The proposed ban is based on the elaborate report by the Kokate Committee submitted in February 2016. The Report deals with more than 3,000 FDCs, and its decisions are based on parameters such as ‘pharmacokinetic/pharmacodynamic (PK/PD) compatibility’, ‘dosage incompatibility’, ‘probable misuse’ and ‘lack of scientific evidence in PUBMED and Google Scholars’. While PK/PD compatibility and existence of studies constitute scientific evidence, ‘dosage compatibility’ and probability of misuse would depend on the practice followed by physicians and pharmacists. Thus, by bringing these two factors, we find an attempt to place regulatory wisdom on a higher pedestal than medical practice or the practice of pharmacists. When seen together with the Rajya Sabha Report where physician’s feedback was sought by the regulatory authority to prove effectiveness of certain FDCs, one conjectures a greater role of physicians in conceptualising and monitoring FDCs in India.
To an extent, to us, the recent debate on combination drugs in India presents a conflict between ‘what works’ in the actual environment and ‘what ought to work’ based on scientifically validated logic and experiments. In STS and innovation studies, this is not a new debate and has come up again in the recent years with emergence of the discourse on frugal innovation and grassroot innovation (Bhaduri, 2016). Many such innovations take place in the actual environment, away from the ideal settings of ‘controlled scientific experiments’. Recent scholarship on science and technology studies, and decision theory, however, remains reluctant to dismiss the merits of these innovations just because they were developed outside the laboratory settings (Gigerenzer, 2008). Laboratory knowledge, to them, is nothing but a specific kind of local knowledge (Dusek, 2006; Irwin & Wynne, 1996). What is required in such a situation is, perhaps, combination of knowledge. In particular, the knowledge of the clinician needs to be brought into the discussion, which has not been done in an adequate manner. A thorough understanding of the process of making combination drugs from their conceptualisation to distribution would be required before dismissing them as irrational. Indeed, this is where the judiciary has also found it difficult to call them unsafe or irrational when long clinical practice of many of these has not revealed any glaring examples of unsafe consequences. In some cases, regulatory authorities, though, have asked for clinical data on FDCs. However, the same point applies here. If long-term clinical practice using these drugs has not revealed any major instances of safety hazards, then whether incurring the risks of clinical trials by the participants is fair from a public health perspective. 24 We, however, could not obtain opinion from the physicians in a large scale about their views on combination drugs. Such a study, however, would have to control for the possible nexus among industry, regulatory agency and the physicians.
Conclusion
Clearly, Indian regulation-making process for new drugs is at a nascent stage and seems to draw heavily on regulatory knowledge of other more industrialised countries. Today, many exemptions from human-based trials are granted if the drugs are available and marketed in advanced economies. 25 Such dependence might, at times, be welcomed from the perspectives of public health and trade, as it reduces duplication of human trial and costs and time of drug approval. The indiscrete dependence on other countries’ regulatory wisdom, however, can put an obstacle to the generation of new knowledge. The Rajya Sabha Report in 2012, sought to delegitimise certain FDCs, primarily, if not solely, because they were not approved in the countries with advanced regulatory capacity, overlooking the possibility of their therapeutic efficacy, which for certain drugs got proven by academic study, subsequently. Such an approach of exclusively relying on other countries’ regulatory data, to legitimise all forms of local practice, perhaps, treats regulatory science as universal, which is clearly at odds with the findings of the scholarship on this issue.
The role of ‘clinical wisdom’ has perhaps not been given due attention in the regulatory processes in India. In their attempts to delegitimise several FDCs, we find an attempt by the regulatory authority to control ‘clinician’s’ practice by banning some FDCs on grounds of their ‘possible misuse’ and ‘overdose’. Given that it is the physicians’ authority to determine use and dosages, such an approach tends to discount the knowledge and autonomy of physicians and puts regulator’s knowledge at a higher pedestal than physicians’ knowledge.
To offer a fresh insight into this issue, we draw upon the growing body of scholarship in the field of regulation making, which often contests the higher prestige of laboratory knowledge. Essentially, this view hints at the debate between ‘what works’ and ‘what ought to work’ (Bhaduri, 2016). More academic studies focusing not only on interest but also on knowledge, values and motivations of entities involved in regulation-making process would certainly be useful to throw more light on these issues.
Inadequate ‘downward causation’ of the rules or thinking of the ‘authority’ seems to be another important feature of Indian new drug approval mechanism. This phenomenon is manifest in, the reason for the persistence of the so-called ‘4-year clause’, which was, perhaps, an appropriate response to inadequate, and slow-moving technological capacity to standardise test assays for new drugs. It has, however, remained a ‘black box’ among the policymakers. What it signals is the deliberations in the regulatory bodies quite often remain un-codified, making their diffusion within the organisation difficult. Informal, or verbal flow of knowledge and information in such cases are important channel of the necessary ‘downward causation’ of decisions and thinking of the ‘authority’ for stability of rules. Ironically, what we find are the rules pertaining to the 4-year clause seems to have derived its stability from non-familiarity of the reason for its adoptions, and not because of its well-entrenched understanding within the members of the regulatory organisation. The downward causation of a ‘decision’ or ‘norm’, therefore, does not necessarily imply spread of its underlying reasons, ironically, even in a science-based industry.
One may also find a separation of the regulatory process pertaining to new drug approval from the larger institutional settings. Apparently, the ‘4-year clause’ can be seen as an attempt, veiled through, to circumvent the regional units from licencing certain kinds of drugs. This issue arises, allegedly, due to competition between regions (individual states), and ‘nexus’ among state regulatory authorities, local industry and physicians. However, if the Report of The Parliamentary Standing Committee on Health and Family Welfare is taken seriously, such nexus exists at the central level too. The regulatory competition between individual states is also not unique to India. In addition, health care in India is the responsibility of individual states making it imperative to put more attention to develop regulatory capacity at the state level.
Footnotes
Acknowledgements
The data for the article were collected as part of a research project sponsored by The French National Agency for Research on AIDSThe French National Agency for Research on AIDS (ANRS), France (ANRS 12213 on Clinical data, bioequivalence and generic drugs). We are grateful to Amit Shovon Ray, the Principal Investigator of this project, for not only sharing his insights on this issue, but also giving his concurrence to use some of the findings of the project for this article. We also acknowledge the active engagement of Brijesh Regal with our research, and the comments we received from Rahul Mongia and the reviewer of the journal.