
Abstract
Cornelia de Lange syndrome (CdLS) is a rare genetic disorder (1 in 10,000–30,000 births) often linked to gastrointestinal (GI) complications such as perforation. The most common GI complication in CdLS is gastroesophageal reflux, caused by improper valve function between the stomach and esophagus, allowing stomach acid to flow back. Mutations are usually sporadic and not inherited. Forensic awareness is vital to avoid misinterpretation as abuse, account for intellectual disability and consent issues, and recognize risks of sudden death from undiagnosed malformations such as intestinal obstruction. At autopsy, features such as facial anomalies, limb defects, and GI pathology can indicate CdLS as a factor in unexplained deaths or related conditions. Given its genetic complexity and potential mosaicism, careful evaluation, even postmortem, is essential for accurate cause of death determination. We describe a case of a 25-year-old female with genetically confirmed lifelong intellectual disability who presented with sudden abdominal pain, vomiting, and melena, followed by unconsciousness. She was declared dead at a tertiary care hospital. Autopsy findings revealed ileal dilatation with perforation. CdLS is a rare genetic disorder with drastic involvement in the GI system. Thus, prompt inquiry of case details and investigations, coupled with a thorough post mortem examination is of paramount importance in such cases.
Introduction
Growth retardation, characteristic facial dysmorphism, primordial short stature, psychomotor delay, behavioral issues, hirsutism, and upper limb reduction defects ranging from mild phalangeal abnormalities to oligodactyly are the hallmarks of Cornelia De Lange syndrome (CdLS), a multisystem developmental disorder. 1 This disorder was also known as Amsterdam dwarfism or Brachymann syndrome. 2 DNA testing is helpful for confirmation of the clinical diagnosis, but the sensitivity is only 50% for mutations associated with the Nipped B-like protein (NIPBL) gene. 3 Its incidence has been estimated to be at 1 in 10,000 to 30,000.
Case Details
A 25-year-old female, mentally disabled since birth with autistic features, had a history of behavioral disturbances from the age of 12 years, for which she received treatment in Bengaluru. She was officially certified by the Government of Karnataka as having a severe intellectual disability with 90% impairment. In 2016, she was diagnosed with hypothyroidism, paralytic ileus, and ascites. Genetic testing later confirmed CdLS.
In early 2024, she was admitted as an inpatient to a rehabilitation home, where she remained for two months. On the evening before her death, she had dinner around 8:30
Ethical Consideration
Ethical approval for this case report was not required as per the guidelines of our institution for individual case studies and was waived by the Institutional Review Board. Written informed consent was obtained from the patient’s next-of-kin for the publication of this case report and any accompanying images or data. All identifying information has been removed from the case report to protect the patient’s privacy.
External Examination
On examination, thick eyelashes, synophrys, a short upturned nose, abdominal distension, small hands (micromelia), and hallux valgus were observed over the body, which was suggestive of features of CdLS (Figure 1).
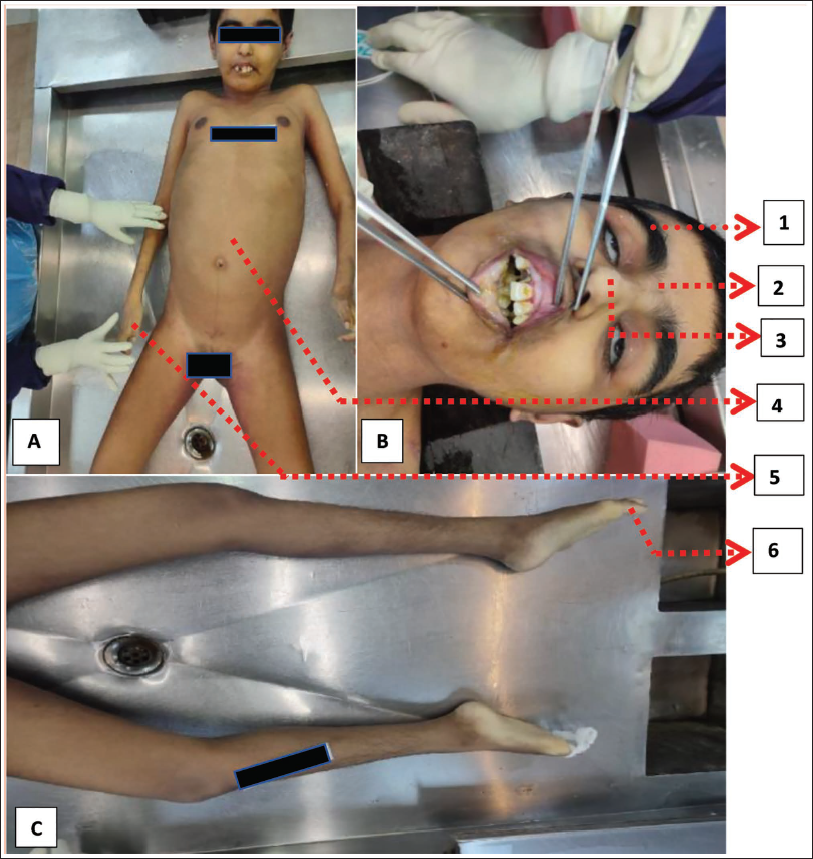
Images A and B show the following external features:
Thick eyelashes
Internal Examination
The crucial positive findings noted in the internal examination involving the small intestinal mucosa (Ileum) were ileal distension, ischemic enteritis, and ileal perforation (Figures 2, 3, and 4, respectively).
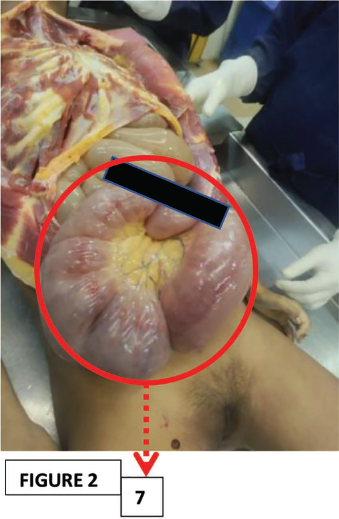
There was a presence of an ileal perforation, measuring 3.5 × 1.5 cm, situated at a point 90 cm from the appendix.
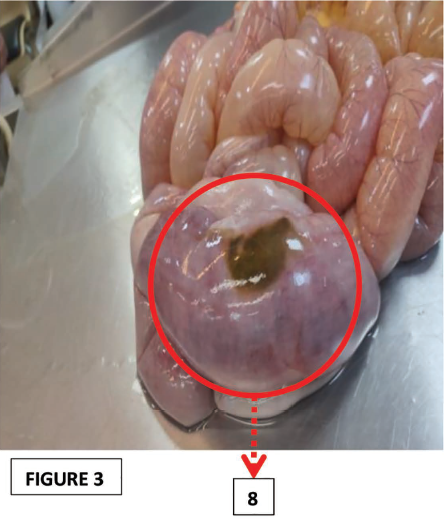
8. Ischemic Enteritis
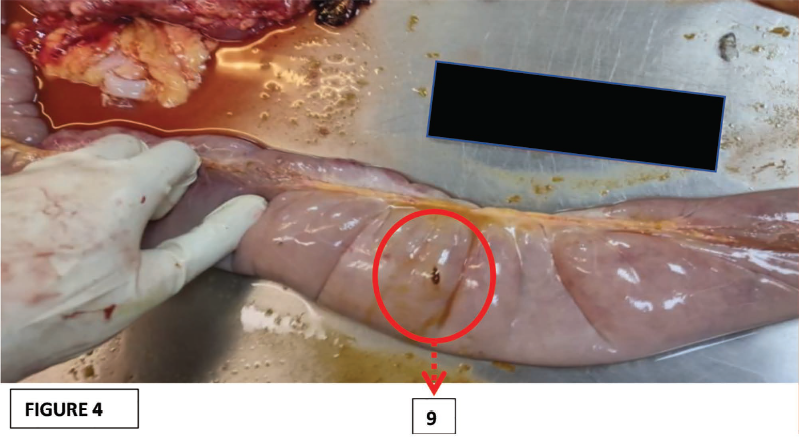
9. Ileal perforation
Histopathological Examination (Figures 5 and 6)
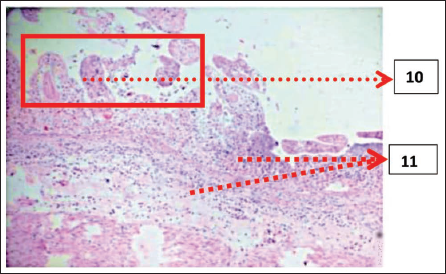
10. Aerated epithelium
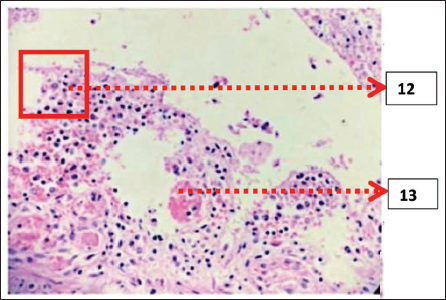
12. Necrosis
Cause of Death
The cause of death was ischemic enteritis secondary to peritonitis.
The postmortem examination of individuals with CdLS must be conducted with the utmost respect for the dignity of the deceased and sensitivity toward the family. Recognizing that CdLS is a rare genetic disorder associated with distinctive physical and medical complications, forensic and pathological evaluation should balance the pursuit of scientific and medicolegal clarity with compassion.
Discussion
The scoring system of CdLS is as follows. 4
The cardinal features mainly include the head and neck features:
Eye manifestations such as synophrys (meeting of the medial eyebrows in the midline) and thick eyebrows, a short nose, concave nasal ridge, upturned nasal tip, and downturned corners of the mouth.
The suggestive features include short fifth finger, small hands and feet, microcephaly, abnormally increased hair growth, prenatal and postnatal growth retardation (<2 standard deviations), hirsutism, global developmental delay, and intellectual disability.
Classic CdLS: >11 points (at least three cardinal features).
Non-classic CdLS: 9 or 10 points (at least two cardinal features).
Molecular testing: 4–8 points (at least one cardinal feature).
Insufficient molecular testing: <4 points.
In the above case, from the aforementioned features, a score of 19 points suggests classic CdLS.
Unique facial dysmorphism, primordial short height, hirsutism, upper limb reduction deficits, unique craniofacial traits, and low Intelligence quotient ranges are the hallmarks of CdLS. 5
A mutation in the NIPBL gene is typical of traditional CdLS. 6
Short stature, microcephaly, low frontal hairline, thick eyebrows, synophrys, long eyelashes, concave nasal root, shallow fossa, thin upper lip, lowered corners of the mouth, widely spaced teeth, micrognathia, small hands, short fifth toe, tiny feet, hirsutism, or intellectual disability are the following. 7
Facial dysmorphic characteristics include hirsutism, intellectual impairment, small hands, clinodactyly, tiny feet, synophrys, thick eyebrows, long eyelashes, shallow fossa, thin upper lip, and lower corners of the mouth. 8
Among the gastrointestinal (GI) malformations in association with this syndrome, a recent study showed that, although sigmoid volvulus is well-known in adults, it is a rare but potentially fatal illness in children. Sigmoid volvulus may therefore be an under-recognized cause of intestinal blockage and digestive perforation that results in death in patients with CdLS, despite the fact that it is difficult to identify. 8
The importance of recognizing the syndromic condition in forensic autopsy is that since this disorder has distinctive dysmorphic features, it may guide the investigators to check missing persons reports for individuals with CdLS. Many of these deaths are related to natural complications. Without recognizing the syndrome, these deaths could be misinterpreted as neglect, abuse, or accidental choking. This helps the forensic experts to correctly attribute the death to natural disease processes, avoiding wrongful legal conclusions.
Confirming CdLS at autopsy allows genetic counseling for surviving family members and any identification of recurrence risks. The relevance of CdLS in forensic medicine lies in the distinct physical features and developmental delays associated with the condition, which can aid in differentiating individuals, estimating age, and identifying victims. Forensic experts may consider these traits as supportive evidence in establishing identity and assessing injuries within the broader context of an investigation. Forensic recognition of CdLS contributes to medical knowledge by documenting natural histories and complications. This helps the public health and research bodies understand mortality trends in rare diseases, which may improve medical care. 8
Conclusion
The autopsy can serve as a confirmatory tool because it allows systematic documentation of external dysmorphic features, internal malformations, and histopathological or genetic findings that match the syndrome. The importance of documenting this rare syndrome in forensic practices is that it clarifies the cause and manner of death by preventing wrongful allegations of neglect, homicide, or abuse. Documented CdLS cases highlight the vulnerability of affected individuals, and they help to establish standards of care for the institutions and caregivers. Since this is an autosomal dominant disorder, the genes affected can be passed on to the next generation. The family members must be counseled. The most common genetic mutation involved is NIPBL (60%). Because of its effects on a person’s health, development, and general well-being, CdLS has medicolegal significance and may give rise to intricate legal issues. The characteristics of the syndrome, such as intellectual disability, developmental delays, and a variety of physical deformities, might affect choices about genetic counseling, healthcare, and disability rights.
Recommendations
In the following pregnancy, parents should receive prenatal diagnosis counseling. Samples from chorionic villous sampling, amniocentesis, or embryonic cells obtained by in vitro fertilization can all be used for prenatal molecular testing.
For areas without access to full gene panels or sophisticated sequencing techniques, targeted single-gene testing of frequently implicated genes, such as NIPBL, is nevertheless useful.
Footnotes
Abbreviations
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical Approval
Not applicable.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Patient Consent
Written informed consent was obtained from the patient’s next-of-kin.