
Abstract
Understanding hydrogen-polymer interactions is essential for designing lightweight and durable hydrogen storage systems for future, sustainable mobility. This study applies molecular dynamics simulations to examine hydrogen solubility, diffusion, permeation and the resulting mechanical response in two representative thermoplastic polymers: polyethylene (PE) and polyamide 6 (PA6). Hydrogen uptake was determined through Grand Canonical Monte Carlo methods, while diffusion coefficients were derived from a mean-squared-displacement analysis. Permeability coefficients were obtained as the product of solubility and diffusivity according to the literature. To assess the mechanical behavior, uniaxial tensile tests were simulated on an atomic scale, under various pressures with and without dissolved hydrogen. The results show that the amorphous regions of PE exhibit a permeability coefficient approximately one order of magnitude higher than that of the amorphous regions of PA6. The semi-crystalline nature of polymers was considered by applying an analytical correction. The corrected permeability values align well with the experimentally measured data from the literature with deviations between -34 % and +24 % for PE and PA6, respectively. In a hydrogen-saturated state, both polymers reveal a marked hydrogen-induced reduction in mechanical response, with PE losing up to 75 % and PA6 up to 85 % of their predicted stiffness under elevated hydrogen pressures and tensile loading conditions. The findings provide molecular-level insights into hydrogen-induced mechanisms in polymers which show high potential to be used in thermoplastic composites for the next generation of Type V hydrogen vessels.
Keywords
Introduction
The field of cargo and individual transportation is currently undergoing disruptive changes to reduce greenhouse gas emissions to a minimum and become carbon neutral within the next decades. Accordingly, the huge developments and technical advances in battery electric vehicles for individual transportation are undebatable. Heavy duty transportation of cargo by trucks is, however, an even more challenging field due to long distances and fast refilling/loading requirements.1–5 Consequently, several research institutions are working on the development and optimization of transportable, lightweight and, most importantly, safe storage systems for hydrogen (H2), as shown in recent studies.6,7
Polymers have been identified in previous studies to be promising alternatives to common metallic solutions due to their addressed insensitivity to hydrogen embrittlement.8–10 In particular, thermoplastic polymers and their composites combine low density with rapid processability and high design flexibility, enabling the fabrication of complex and lightweight pressure vessels for H2 storage applications.1,3,11 These advantages make them attractive candidates for matrix materials in next-generation Type V tanks. Nevertheless, the small molecular size and high diffusivity of hydrogen facilitate permeation through the polymer matrix, which can lead to gas losses, material degradation, and safety concerns under high-pressure operation. Common liner materials like polyethylene (PE) and polyamide 6 (PA6) have already justified their application in hydrogen applications, which makes them attractive for new, linerless storage solutions, e.g., Type V pressure vessels. Short cycle times, increased toughness and improved recyclability compared to common thermoset matrices emphasize the potential of PE and PA6 in structural applications when it comes to hydrogen storage.1,9,11
In current hydrogen storage technologies, polymers are typically part of fiber-reinforced composite systems.3,11 In state-of-the-art lightweight hydrogen storage solutions, like Type IV pressure vessels, thermosetting matrices, as epoxy resins, are combined with continuous carbon fibers to provide structural integrity, while a separate polymer liner ensures gas tightness. In contrast, emerging linerless Type V concepts aim to utilize thermoplastic polymers as both structural matrix and barrier material. In these systems, continuous fiber reinforcements remain essential for load-bearing capacity. Nevertheless, molecular transport is primarily governed by the polymer matrix itself.9,12–16 Therefore, this study focuses on the polymer matrix to isolate intrinsic hydrogen-polymer interactions before addressing more complex composite architectures.
Hydrogen transport through polymers is commonly described by the solution–diffusion model, in which molecular hydrogen first dissolves in the amorphous regions of the polymer and subsequently diffuses through interconnected free-volume pathways.9,12–16 The resulting permeability, P, is defined as the product of solubility, S, and diffusivity, D, of the corresponding material. These parameters are sensitive to factors such as temperature, pressure, polymer morphology, and chain mobility. In semi-crystalline polymers, like PA6 and PE, the crystalline domains are commonly considered as impermeable barriers, while the amorphous regions serve as the primary transport region.9,10,14–22 Consequently, the overall permeability depends not only on intrinsic molecular interactions but also on the tortuosity imposed by the crystalline domains and the degree of crystallinity.14,23–25
Over the past decades, experimental studies have generated data on hydrogen permeation in various polymer systems.12,13,26 Nevertheless, separating the individual contributions of solubility and diffusion, as well as accessing molecular-level mechanisms, remains challenging. Furthermore, measuring hydrogen uptake and transport at elevated pressures is experimentally demanding, particularly when decoupling physical sorption from chemical effects. Molecular dynamics (MD) simulations have gained significant relevance as a complementary method for investigating polymer–hydrogen interactions at the atomistic scale.9,20,27,28 Using validated force fields, solubility coefficients can be determined from equilibrium adsorption behavior obtained through Grand Canonical Monte Carlo (GCMC) simulations, while diffusion coefficients can be derived from mean-squared displacement (MSD) analysis.18,20,29,30
Beyond permeation, the presence of dissolved hydrogen can significantly influence the mechanical response of polymers.8,31,32 Hydrogen molecules occupying free-volume sites can alter stiffness, yield behavior, and ductility by modifying local segmental mobility and intermolecular interactions. Experimental results reported in the literature show varying trends, including contradictory observations for thermoplastic materials: Menon et al. 31 reported that exposure to high-pressure hydrogen environments (70-100 MPa) led to a moderate stiffening in semi-crystalline thermoplastics, which was attributed to chain alignment and densification under gas dissolution. In contrast, Alvine et al. 32 observed a reduction in tensile strength of high density polyethylene under hydrogen pressures up to 42 MPa, indicating a softening effect at pressures over 28 MPa. Consequently, these studies demonstrate that the mechanical response of polymers to hydrogen is strongly material- and morphology dependent, potentially shifting from stiffening to softening behavior. Understanding these contrasting results is essential for predicting the structural integrity and long-term durability of polymeric liners and composite structures in Type V hydrogen storage vessels.
Within the framework of a digital materials engineering process and the aforementioned state-of-the-art, this study employs atomistic molecular dynamics simulations to investigate hydrogen solubility, diffusion, and mechanical behavior in PA6 and PE as representative thermoplastic polymers for future hydrogen storage applications. Amorphous molecular models of the investigated materials were generated and equilibrated utilizing the commercial software package Simcenter Culgi (Siemens Digital Industries Software, Plano, Texas, USA). Subsequently, GCMC simulations were performed to determine hydrogen uptake and MSD analyses to derive diffusion coefficients. Permeability coefficients were obtained from the product of solubility and diffusivity and corrected for crystallinity to enable comparison with experimental data. Finally, uniaxial tensile simulations were performed at different pressures and associated H2 concentrations to assess the influence of dissolved hydrogen on mechanical behavior. The obtained results aim to (i) quantify the molecular-scale transport properties of hydrogen in PA6 and PE and (ii) analyze the influence of hydrogen uptake on the mechanical response of these thermoplastic polymers by means of atomistic molecular dynamics simulations. The study provides molecular-level insight into hydrogen–polymer interactions and contributes to a better understanding of how polymer morphology and hydrogen solubility affect permeability and mechanical performance in potential hydrogen storage applications.
Materials and methods
The following paragraphs provide detailed information on the two investigated polymers polyethylene (PE) and polyamide 6 (PA6) and how the modeling was conducted from a molecular dynamics perspective. To keep the modelling and computational effort reasonable, only unfilled polymers were considered within this study. Furthermore, the simulation schemes applied and the boundary conditions for the calculation of the H2 solubility and diffusivity will be explained before the setup of the atomistically modeled tensile test approach in a saturated H2 atmosphere is described.
Polyethylene and polyamide
Table 1 indicates the high variability of PE and PA6 grades regarding molecular weight, Tg and crystallinity. In the following, these three parameters were kept constant for both polymers to generate comparable results. The molecular weights of the analyzed PE and PA6 were set to 2.5 × 104 g/mol and 2.4 × 104 g/mol, respectively, while the crystallinity values were 77.5 wt% and 25 wt%. The glass transition temperatures were derived from the molecular model and used to assess the plausibility of the generated representative units of the polymers.
Derived from the inherent chemical structure, PE is a non-polar polymer, characterized by comparatively high chain flexibility and weak intermolecular interactions dominated by van der Waals forces. In contrast, PA6 is a polar polymer, which is capable of hydrogen bonding. This leads to a stronger intermolecular cohesion and therefore different barrier and mechanical properties. This contrast makes PE and PA6 suitable comparative model systems for studying the influence of polymer structure and intermolecular interactions on hydrogen permeation and hydrogen-induced changes in mechanical response.
Molecular dynamic simulations
Atomistic polymer chains were generated in Simcenter Culgi (Siemens Digital Industries Software, Plano, Texas, USA) using the corresponding repeating units of PE and PA6 from Table 1. Due to the complexity and computational effort required to mimic crystalline structures in the polymer network, the authors decided to perform all molecular dynamics (MD) simulations on representative amorphous regions. The amorphous models comprised three PE chains with 850 repeat units and four PA6 chains with 200 repeat units each. The resulting structures were arranged into cubic mesoboxes, with a side length of approximately 5.3 nm after relaxation and energy minimization. For the following permeability considerations, only the amorphous morphology of each polymer was analyzed, whereas the crystalline domains were assumed to be impermeable to hydrogen.
All MD simulations performed in this study employed the DREIDING 39 force field with full atomistic resolution, which includes directional hydrogen-bonding terms to account for polar interactions in PA6. Temperature and pressure were controlled using the Nose–Hoover thermostat and Andersen barostat, respectively. These were chosen according to the recommended guidelines in the software documentation. 40 Nonbonded interactions were truncated at 12.5 Å, and periodic boundary conditions were applied in all three spatial directions within the chosen Cartesian coordinate system. Energy minimization was performed using the default settings of the software to remove atomic overlaps. Subsequently, a thermal annealing protocol was applied, cycling between 200 K and 500 K in increments of 50 K with 50 ps per step for two full cycles. This was followed by equilibration runs in the canonical and isothermal-isobaric ensembles to ensure structural relaxation. 40 The timestep was set to 0.5 fs for the equilibration and relaxation runs. The generated, relaxed mesoboxes served as the starting configuration for all subsequent MD simulations.
To assess the accuracy of the molecular models in the initial state, the predicted densities, ρ, and glass transition temperatures, Tg, of both polymers were compared with experimental literature data. In this work, standardized procedures according to the literature were followed.9,18,40,41 For both parameters and materials, the maximum deviations are within a range of ±15 % with respect to the values provided in Table 1. Consequently, evidence of the validity of the mesoboxes shown in Figure 1 is given. (a) Representation of the valid PE mesobox with a density of ρPE = 805 kg/m3 and a glass transition temperature of Tg,PE = 227 K; (b) Representation of the valid PA6 mesobox with a density of ρPA6 = 1004 kg/m3 and a glass transition temperature of Tg,PA6 = 322 K.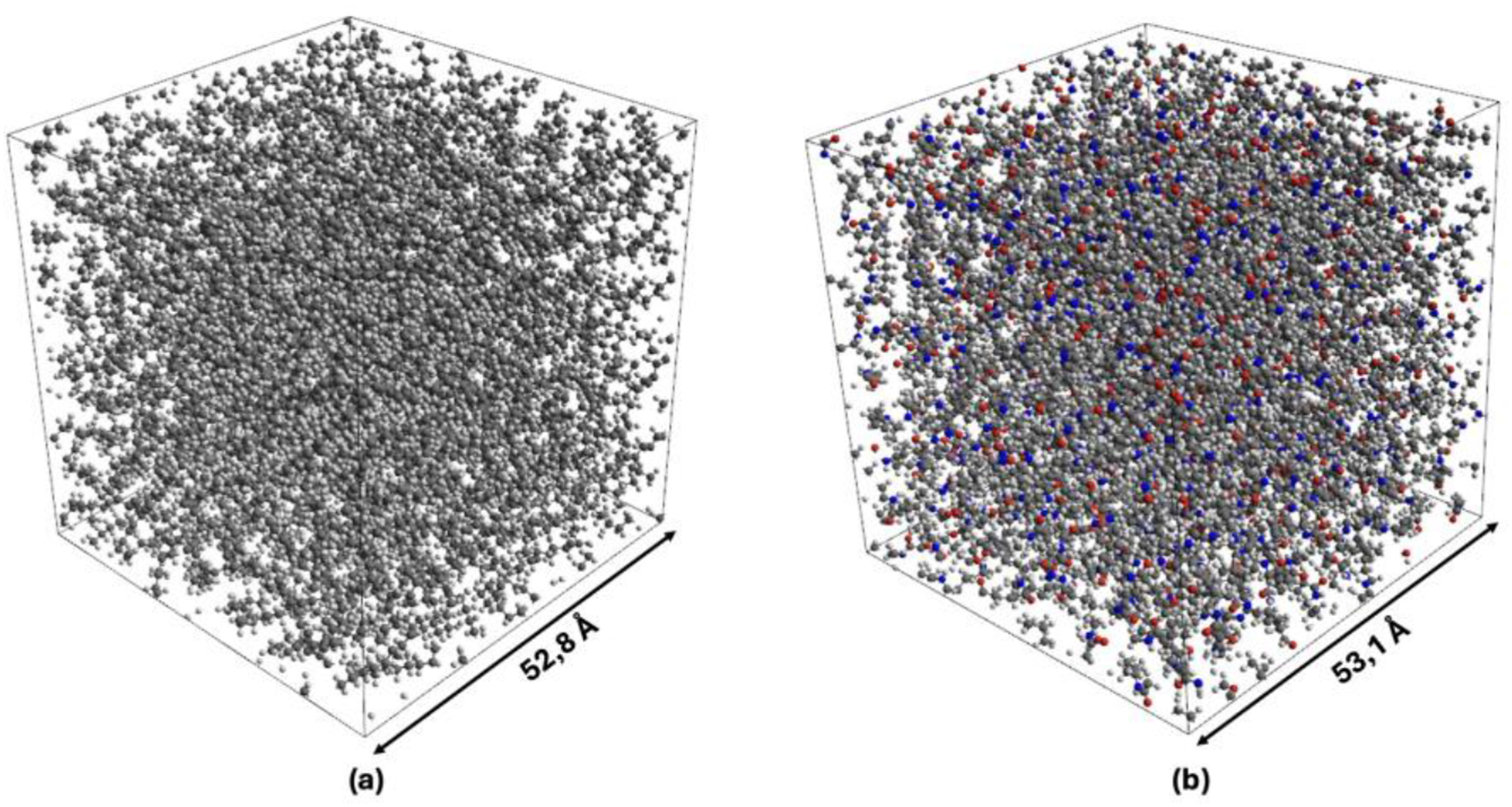
Solubility and diffusion simulations
The solubility of a gas in a polymer quantifies the amount of gas dissolved by the polymer network at a given temperature and pressure.9,14,42 According to Henry’s law, it is defined as the ratio of the dissolved concentration, C, to the fugacity, f, in the dilute limit:
As the applied pressures within this study did not exceed 1 MPa, hydrogen was treated as an ideal gas, allowing that f can be approximated by the pressure, p. This assumption (f ≈ p) allows an easier interpretation of the latter results from an engineering perspective.
GCMC simulations were performed to determine the solubility coefficient, S, for hydrogen in PE and PA6. In this method, the pressure is externally imposed, and hydrogen molecules are probabilistically inserted and removed from the mesobox to sample equilibrium states.20,40,41,43 Simulations were conducted at pressures up to 1 MPa, yielding adsorption isotherms in the low-pressure regime from which S was obtained as the slope.
The solubility is reported in cm3(STP)/cm3 MPa, where STP denotes the standard temperature and pressure reference of 273.15 K and 1 bar.
To compare the simulated results with experimental data from semi-crystalline polymers, the simulated solubility coefficients in the amorphous regions, S∗, were corrected for the degree of crystallinity, α, by using a simple linear relation according to Klopffer et al.
13
:
The diffusion coefficient, D, is the second transport parameter required to calculate the permeability, P. D was obtained by MD simulations based on an MSD analysis. For a system containing N hydrogen molecules, the MSD is defined as27,40,44:

A double logarithmic representation of the MSD over time allows the identification of the diffusion regime.9,20,44 For Fickian diffusion, the slope in the corresponding plot is approximately equal to one 
β is a geometric correction factor in this case and is equal to 2/3, following the recommendations in Su et al. 9 This approach accounts for the reduced connectivity of amorphous domains within semi-crystalline polymers.
Molecular tensile tests
To investigate the effect of dissolved hydrogen on the stress-strain behavior of PE and PA6, uniaxial tensile tests on a molecular scale were performed on both, hydrogen-free and hydrogen-saturated models. The saturated models were generated by using the aforementioned approach of GCMC simulations at distinct pressure levels, followed by equilibration to ensure homogeneous gas distribution within the matrix. All simulations, with and without H2, were performed at a constant temperature of 296 K and four pressures of 1 bar, 10 bar, 100 bar and 350 bar. The fully atomistically modeled mesobox of PE and PA6, with an initial side length of 53.1 Å, was uniaxially deformed by an enforced displacement boundary condition along the x-axis. A constant strain rate of 109 s−1, which is very high but typical for MD simulations,45,46 was applied until a maximum strain of 30 % was reached. The resulting stress, σx, and strain, εx, were derived from the virial stress tensor and the box deformation at each timestep increment. As an example, the initial and deformed mesobox of PA6 at 1 bar is illustrated in Figure 2(a) and (b). (a) Representation of the initial PA6 mesobox with side lengths of lx = ly = lz = 53.1 Å before deformation; (b) Representation of the deformed PA6 mesobox at 30 % engineering strain, corresponding to an elongated x-dimension of lx = 69.0 Å and a reduced transverse dimension of ly = lz = 47.0 Å due to the Poisson effect. Colour coding of atoms: dark gray - carbon (C), light gray - hydrogen (H), red - oxygen (O) and blue - nitrogen (N).
Due to thermal fluctuations and the finite size of the atomistic models, instantaneous stresses exhibit high frequency oscillations. 47 To extract the underlying mechanical response, the stress–strain data were smoothed using a moving average filter, effectively reducing molecular-scale noise while preserving the physical deformation trend. This approach enables a direct comparison of the elastic behavior between hydrogen-free and hydrogen-saturated systems. Analogously to the plausibility check of the solubility and diffusivity properties, the mechanical response of the hydrogen-free systems was validated against experimental reference data from the open access database CAMPUS - Computer Aided Material Preselection by Uniform Standards (Chemie Wirtschaftsförderungs-GmbH, Frankfurt, Germany). Although the applied methodology provides valuable insights into the hydrogen-induced changes in polymer mechanics, several model-related limitations must be considered. The models represent fully amorphous systems without explicit crystalline phases or fiber interfaces. Furthermore, the applied strain rate is several orders of magnitude higher than in macroscopic experiments, and the finite simulation cell size limits the accessible strain and relaxation timescales. Future research will focus on these limitations to quantify the sensitivity of the simulations with respect to the differences in the aforementioned parameters.
Results and discussion
The following subsections will provide all results obtained from molecular dynamics simulations with respect to permeation and tensile properties of the investigated PE and PA6 polymers. Furthermore, all derived parameters were put into perspective of peer-reviewed experimental literature.
Hydrogen permeation through polyethylene and polyamide
According to equation (1), the solubility of a material is defined as the change in concentration over the change in pressure (or fugacity above 1 MPa). Consequently, the slopes of the calculated adsorption isotherms in Figure 3(a) are used to derive the solubility of PE and PA6. The linear regression of both polymers predicts a more than doubled solubility in the amorphous regions of PE with (a) Simulated adsorption isotherms of PE and PA6 at room temperature (RT). The slope of the linear regression provides the solubility of the polymer with respect to standard temperature and pressure (STP) conditions; (b) Comparison of the simulated amorphous, for semi-crystallinity corrected (see equation (2)) and from literature obtained solubility values.
In terms of diffusivity, the development of the mean squared displacement, MSD, over time provided the basis for deriving the corresponding parameter. The diffusivity, D*, is extracted from the region of Fickian diffusion, which is equivalent to a slope equal to 1, by applying equation (4). From the line plot in Figure 4(a), one can observe that in the amorphous state, the MSD of PE is about one order of magnitude higher compared to PA6. This can be associated with a higher diffusivity in these regions. Due to the semi-crystalline nature of both polymers, again an analytical correction was applied based on equation (5). This relation is derived from the assumption of zero diffusion in crystalline regions. As depicted in Figure 4(b), the impact of crystallinity is even more pronounced in terms of diffusivity than previously presented for solubility. In total, the corrected diffusivity, D, for PE is roughly twice as high as that of PA6 by (a) Line plots of the mean squared displacement over time development for PE and PA6. Within the Fickian diffusion region, a linear regression was performed which is indicated by the dashed lines. Note that the gradient triangles are illustrated schematically and not in scale; (b) Comparison of the simulated amorphous, for semi-crystallinity corrected (see equation (5)) and from literature obtained diffusivity values.
In a subsequent and final step, the overall hydrogen permeability, P, of the investigated polymers was calculated by multiplying the solubility and diffusivity, (a) Visual comparison of the simulated amorphous, for semi-crystallinity corrected and from the literature taken permeability values in a bar plot; (b) Tabular listing of permeability values determined for PE and PA6 within this study.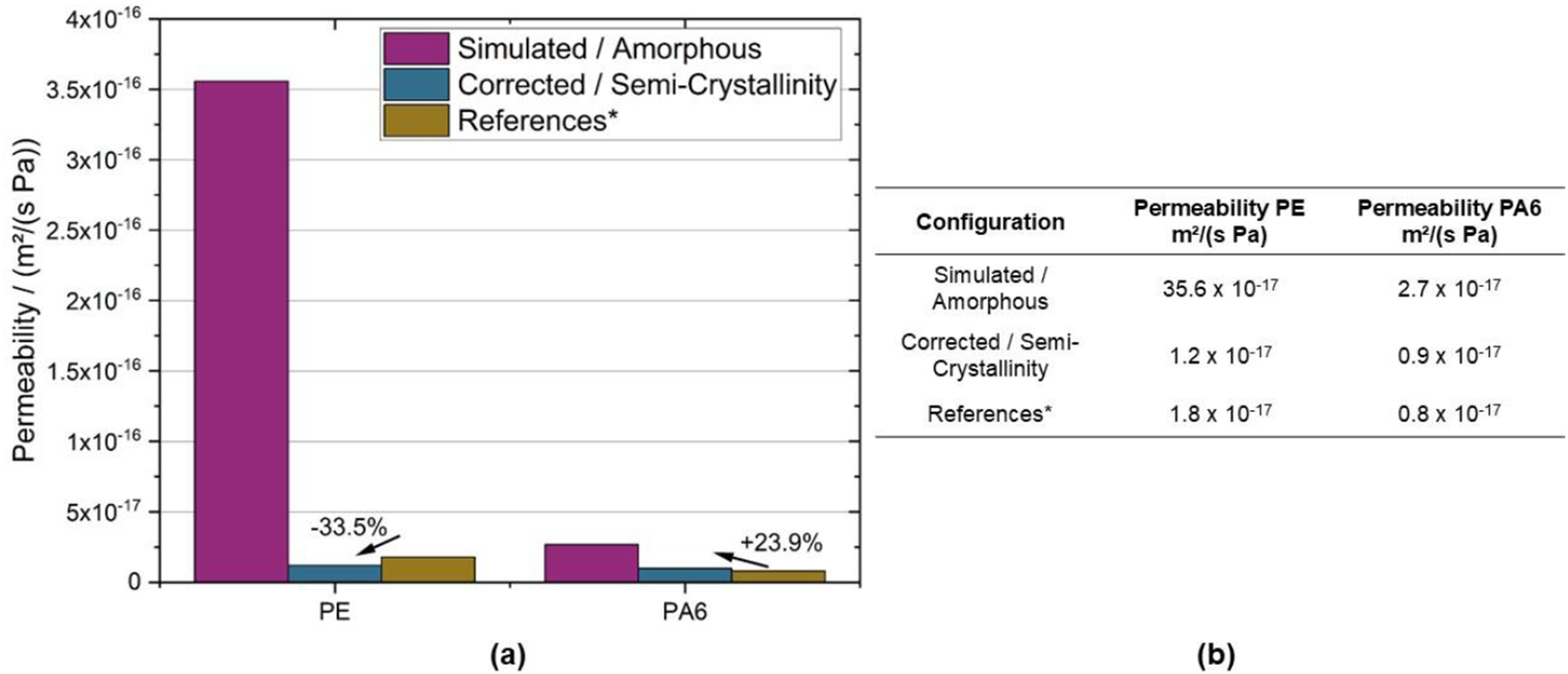
The permeability regarding H2 is drastically higher in the amorphous PE regions. Solely the degree of crystallinity in polyethylene dominates the hydrogen permeability of this polymer. In contrast, this effect is less pronounced in PA6. Due to the chemical structure of polyamide, the formation of crystalline regions is much more limited than in PE. Furthermore, the more complex chemical structure is the reason for a lower permeability in the amorphous state as indicated by the corresponding magenta-colored bar in the chart. This underlines the importance of the (super)molecular morphology in polymers as a key parameter when it comes to tailored permeability properties in the case of H2 storage. The relatively small deviations between the simulated permeability values and their experimental counterparts from the literature provides evidence for the accuracy of the simulations. Although the exact chemical formulation of the polymers would be required to generate more accurate data or material properties, this information is commonly unknown.
Mechanical tensile properties of polyethylene and polyamide in hydrogen atmosphere
From a mechanical perspective, the influence of dissolved hydrogen on the mechanical material properties is highly interesting. Tensile tests on an atomic level, with and without H2, were performed to quantify the corresponding impact. Indeed, the simulations revealed a significant softening effect on the tensile properties of PE and PA6. Figure 6 presents the derived stress, σ, over strain, ϵ, curves for both polymers. The curve coloring indicates the applied pressure level while the more transparent curves are associated with the results in H2 atmosphere. As reference, the in red marked σ − ϵ curves at 1 bar and without H2 were considered. In (a) of Figure 6 the resulting curves of PE illustrate a clear hydrogen induced softening effect. The averaged stress level at 30 % strain is reduced to one third of the value in a hydrogen-free environment. This effect can be quantified as well by analyzing the Young's modulus, E, of the PE. At a pressure level of 10 bar, 100 bar and 350 bar, the corresponding E values in H2 atmosphere were reduced by -73 %, -70 % and -75 %, respectively. A similar behavior one observes for PA6, as illustrated in Figure 6(b). The reductions of E, in H2 environment, are even higher for 10 bar and 100 bar with values of -81 % and -85 % compared to PE. At 350 bar a more moderate reduction of -55 % was observed. In absolute numbers the simulated Young's modulus of PE, without hydrogen, matches the data from the literature in Table 1 well, by showing a value of 950 MPa. However, the corresponding value of PA6 is only 50 % of the number shown in Table 1. This might be a consequence of the purely amorphous morphology of the investigated mesobox. (a) Stress-strain curves of PE obtained by atomistic uniaxial tensile loading without and with dissolved H2; (b) Stress-strain curves of PA6 obtained by atomistic uniaxial tensile loading without and with dissolved H2.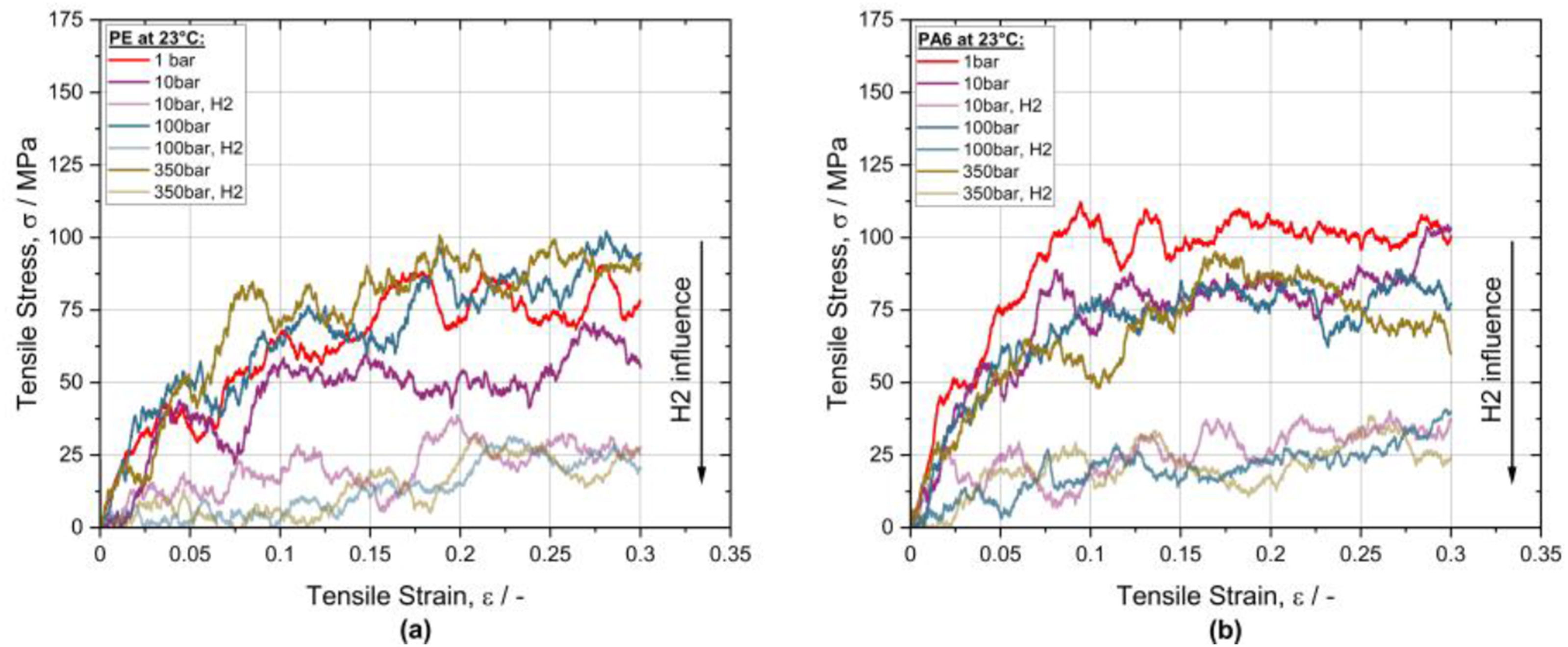
The mesoboxes of both polymers were exposed to a displacement-controlled deformation up to a maximum strain of 30 %.
The observed softening in both polymers suggests that the dissolved hydrogen influences the mechanical response of the polymer matrix. A possible contributing factor is the presence of hydrogen within free-volume sites, which may alter local packing and intermolecular interactions. The simulation temperature of 296 K must be considered in relation to the glass transition temperatures of the investigated polymers (Tg,PE ≈ 227 K and Tg,PA6 ≈ 322 K). Under these conditions, polyethylene is in an entropy elastic/rubbery state, resulting in high chain mobility and ductility. In contrast, polyamide 6 is below its glass transition temperature, leading to reduced segmental mobility and a comparatively stiffer mechanical response. This difference in thermodynamic state is expected to influence both mechanical behavior and hydrogen transport properties, as increased chain mobility facilitates diffusion, whereas the restricted mobility in an energy elastic state (below Tg) may hinder molecular transport. In addition, intermolecular interactions further contribute to the differences between both polymers. Intermolecular interactions in PE are dominated by van der Waals forces, resulting in comparatively high chain flexibility and lower resistance to deformation. In contrast, PA6 exhibits stronger intermolecular cohesion due to hydrogen bonding between amide groups, which reduces chain mobility and increases resistance to deformation. Consequently, both the thermodynamic state of the polymer and the nature of intermolecular interactions are likely to contribute to the observed differences in mechanical response and hydrogen diffusion. Nevertheless, the exact mechanisms governing the pressure-dependent behavior are not fully understood and will be addressed in future research.
Conclusion
The atomistic simulations (GCMC, MSD and tensile simulations) performed in this work showed that this type of modeling can consistently capture how hydrogen is transported through amorphous morphologies of thermoplastic polymers and how the presence of hydrogen affects the mechanical response. The results demonstrated that PE and PA6 exhibit different hydrogen–polymer interactions, confirming that these effects cannot be described by a single material parameter and require a coupled consideration of constituent configuration and final polymer morphology, e.g., crystallinity. The simulation workflow proved capable of resolving the distinctly different solubility and diffusivity levels in amorphous models of PE and PA6, including the order-of-magnitude higher permeability obtained for the amorphous regions of PE, and of capturing the softening observed in the tensile simulations with dissolved hydrogen.
Simultaneously, the results underline the methodological boundaries of fully amorphous atomistic models. Absolute solubility and diffusivity values are affected by finite-size limitations and the analytical crystallinity correction, while the mechanical response is also influenced by the inherently high strain rates required in MD. These limitations reduce the predictive accuracy of absolute values but do not affect the consistency of relative trends.
In conclusion, this study showed that molecular dynamics simulations are well suited for identifying material-dependent trends in hydrogen solubility, diffusivity, permeability and hydrogen-induced softening in polymers. This is essential for early material considerations in the development of high performance applications such as Type V hydrogen storage pressure vessels.
Footnotes
Acknowledgments
The authors would like to thank Siemens Industry Software GmbH for providing the molecular dynamics simulation software Culgi and Prof. Dr. Michael Heiss (Siemens AG Österreich) for his valuable scientific input during bilateral meetings and discussions.
Author contributions
Conceptualization, M.H. and A.K. ; methodology, M.H.; software, M.H.; validation, M.H and A.K.; formal analysis, M.H. and A.K.; investigation, M.H. and A.K.; resources, Z.M.; data curation, M.H. and A.K.; writing—original draft preparation, M.H. and A.K.; writing—review and editing, M.H., A.K. and Z.M.; visualization, M.H. and A.K.; supervision, Z.M.; project administration, Z.M.; funding acquisition, Z.M. All authors have read and agreed to the published version of the manuscript.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was funded by the Austrian Research Promotion Agency (FFG) within the HYPOLFAIL project (![]() and “Mobilitätswende”, grant number FO999924660), the LIFT-C project GH2MOB, and Siemens AG Austria.
and “Mobilitätswende”, grant number FO999924660), the LIFT-C project GH2MOB, and Siemens AG Austria.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data Availability Statement
Processed data underlying the key findings of this study are available in aggregated form from the corresponding author upon reasonable request. Raw molecular dynamics trajectories and simulation files cannot be shared publicly due to software licensing restrictions and file size limitations.