
Abstract
This work introduces a promising biodegradable copolymer synthesized through the fusion of D,L-aspartic acid (ASP) and L-glutamic acid (GA) utilizing melt polymerization. Employing infrared spectroscopy (IR), nuclear magnetic resonance spectroscopy (1H NMR, 13C NMR), and X-ray diffraction (XRD), the copolymer’s structural characterization highlights its distinctive physicochemical attributes. The synthesis, conducted with a facile and controllable melt polymerization method, yielded a remarkable product yield of up to 81%. The optimization of water absorption properties involved a meticulous exploration of various diamine cross-linking agents—hexamethylene diamine (HMD), lysine (LYS), and a synthesized tartaric acid dihydrazide (TD) derivative. Fourier transform infrared spectroscopy (FT-IR), nuclear magnetic resonance spectroscopy (NMR), and scanning electron microscopy (SEM) were employed to confirm the copolymer’s structure, morphology, and cross-linking efficiency. The findings underscore exceptional water retention capabilities, with a peak swelling ratio of 11.874% achieved at a 10% concentration of hexamethylene diamine. Beyond advancing superabsorbent materials, this study contributes to mitigating environmental concerns associated with non-biodegradable alternatives.
Keywords
Introduction
Superabsorbent polymers (SAPs) serve a crucial role in various industries, notably in agriculture, where effective water retention is paramount for sustainable crop production. However, the predominant use of non-biodegradable SAPs, exemplified by acrylate polymers, raises significant environmental concerns. Despite their straightforward synthesis and remarkable water-absorption properties, these polymers persist indefinitely in the soil, contributing to environmental pollution.1–4 In contrast, poly (amino acid) and its copolymers present a promising avenue for sustainable development. Biodegradable, biocompatible, and non-toxic, these polymers can act as soil amendments, enhancing plant production while minimizing soil erosion.5,6
The category of polyamino acids, characterized by peptide bond linkages and reactive side chains containing functional groups such as –COOH and –CONH2, has produced various biodegradable SAP polymers, including poly (aspartic acid) (PASP),7,8 poly (glutamic acid) (PGA), 9 and poly (lysine) (PLYS). 10 The synthesis of PASP can be divided into two different reaction methods: The first is based on the thermal condensation of aspartic acid monomers under vacuum conditions, requiring temperatures between 160 and 260°C for a period of one to 4.5 h with H3PO4 (85%) as a catalyst. The second method involves the use of monomers such as maleic acid and urea with a mixture of phosphoric and sulfuric acid.11–13 To create the superabsorbent polymer, succinimide (PSI) was used as a raw material, and various diamines were employed as cross-linking agents. Tests have shown that this PASP product is capable of absorbing large quantities of water. 14 Meanwhile, PGA, a high molecular weight polymer, can be produced through microbial 15 and chemical processes. 16 Additionally, PGA finds application in the agricultural, pharmaceutical, and chemical industries as a resin with a high water absorption capacity. 17
In this study, our focus is on the development of a new copolymer composed of D,L-aspartic acid (ASP) and L-glutamic acid (GA). Utilizing melt polymerization for its ease of management and high yields of up to 81%, this copolymer aims to amalgamate the distinct properties of PASP and PGA, yielding a material with improved characteristics suitable for a broader range of applications. Our investigation extends beyond synthesis, exploring the effects of catalysts, reaction parameters, and the molar ratio of (ASP) to (GA). The structural characterization of our material involves a comprehensive analysis using techniques such as infrared spectroscopy (IR), Nuclear Magnetic Resonance Spectroscopy (1H NMR, 13C NMR), and X-ray diffraction (XRD).
Additionally, we endeavor to identify optimal conditions for water absorption by poly (aspartic-co-glutamic acid) using various diamine cross-linking agents, including commercially available amino acids hexamethylene diamine (HMD) and lysine (LYS), as well as tartaric acid dihydrazide (TD) synthesized from tartaric acid. The structural integrity, morphology, and efficiency of the cross-linking procedure are meticulously examined through Fourier transform infrared spectroscopy (FT-IR), NMR measurements of T2 relaxation times (for three different times: 0.5, 3, and 8 s), and scanning electron microscopy (SEM).
Experimental section
Materials
This study utilized D,L-Aspartic acid (ASP ≥99%),
Synthesis and optimization
Synthesis of pre-polymer
For the synthesis of Poly (succinimide-co-glutamic acid) (PSI/GA), we used the approach illustrated in Figure 3(a). In a 100 mL round bottom flask, ASP and GA were placed, and then 3 g (0.03 mol) of phosphoric acid (85% w/w) was introduced as a catalyst. The copolymerization took place under specific temperature conditions in a vacuum. After being allowed to cool to room temperature, the product was washed multiple times with distilled water to remove the catalyst and reactants. Subsequently, the copolymer was filtered and ultimately dried at 80°C for a period of 4 h.
Selection of the molar ratio of ASP to GA
Previous studies have indicated that maintaining a higher molar concentration of D,L-aspartic acid than its grafting monomer,
Selection of the molar ratio of ASP to GA.

Selection of reaction time
Selection of the reaction time.

Selection of the reaction temperature
Selection of the reaction temperature.

Crosslinker synthesis
In addition to the readily available commercial crosslinkers HMD and LYS, another crosslinker TD was synthesized in the laboratory by reaction of diethyl tartrate with hydrazide to produce diamino compound.
L-(+)-Tartaric Acid Dihydrazide crosslinker
A mixture of N2H4.H2O (4.9 g, 152 mmol, 80%) and diethyl L-(+)-tartrate (5.15 g, 25 mmol) was stirred for 8 h to make a white precipitate. The precipitate was separated through filtration and washed with Et2O to produce L-(+)-tartaric Acid Dihydrazide (4 g, 89%), with a melting point of 185°C.
18
Figure 1 shows the reaction between diethyl L-(+)-tartrate and hydrazine. Synthesis of tartaric acid dihydrazide (TD).
The synthesis of the tartaric acid dihydrazide (TD) crosslinker was confirmed using 1H-NMR and 13C-NMR spectroscopy (Figure 4).
Crosslinking and hydrolysis to clPASP/GA
PSI/GA’s succinimide ring structure can be easily reacted with an amine group. This allows bifunctional amines to link two of the polymer chains together (Figure 3(b)), forming a hydrogel network (Figure 3(c)). As illustrated in Figure 2, the most commonly used crosslinking agents are hexamethylene diamine (HMD) and lysine (LYS) amino acid, which are available commercially. Additionally, the crosslinking agent based on tartaric acid dihydrazide (TD) was synthesized and used. The choice of crosslinking agent could influence the hydrogel’s swelling and mechanical properties. Diamine crosslinkers used for PASP/GA-based hydrogels.
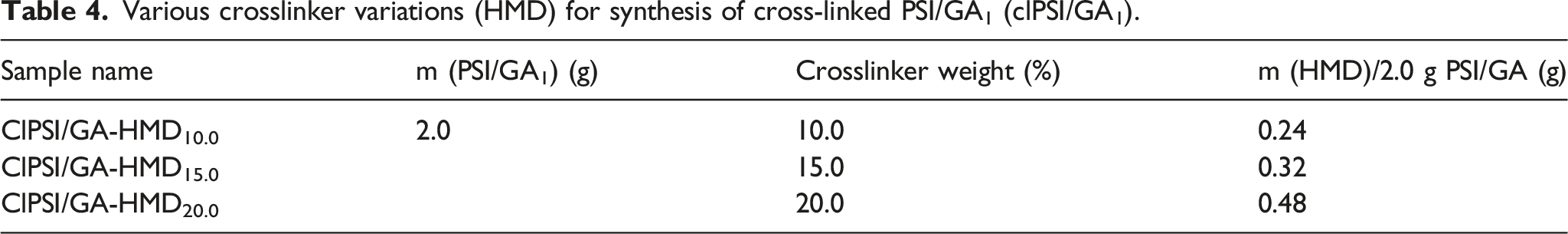
Crosslinking by HMD
Various crosslinker variations (HMD) for synthesis of cross-linked PSI/GA1 (clPSI/GA1).
Crosslinking by LYS
Various crosslinker variations (LYS) for synthesis of cross-linked PSI/GA1 (clPSI/GA1).

With 15.0 and 20.0% crosslinker, no hydrogels were formed.
Crosslinking by TD
Various crosslinker variations (TD) for synthesis of cross-linked PSI/GA1 (clPSI/GA1).

With 10.0 % crosslinker, no hydrogels were formed.
Formation of PASP/GA Hydrogels through the Hydrolysis of clPSI/GA
Hydrolysis of clPSI/GA1 to clPASP/GA1 with NaOH.

Characterization methods
Product yield, X-ray diffractometer, NMR analysis, swelling ratio, FTIR spectroscopy, swelling ratio, and SEM observation were employed for comprehensive characterization.
Determination of the copolymer yield
The purity of the copolymer product can be determined by dissolving it in DMF and using a mass balance to calculate the amount of impurity which does not dissolve.
The product was dissolved in 10 mL DMF and left to stand for 24 h at room temperature. The solution was then filtered through a filter paper, which was dried at 40°C for 3 h until a constant weight was achieved. The purity of the product was then determined using equation.
1
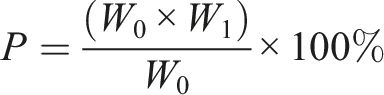
Let W0 and W1 represent the weight of the product and the weight of insoluble materials in filter paper respectively, with p being the purity of the product.
The products had to be washed several times with de-ionized water to eliminate the catalyst and non-reacting components, and then filtered. The copolymers obtained were dried at 80°C for 4 h. The product yield can be calculated using the equation:
2

X-ray diffractometer
The synthesized PSI/GA polymer was characterized by X-ray Diffraction analysis, which was carried out using an X-ray diffractometer (XRD-D8, Advance, Bruker) with a scanning rate of 0.02° per second. The analysis covered a 2θ range from 5° to 80°.
NMR analysis
The 1H NMR and 13C NMR spectra of the PSI/GA and PSI in DMSO, as well as new crosslinkers from TD in D2O, were measured using a 300 MHz (Bruker) spectrometer. Additionally, 1H NMR spectra of PASP/GA hydrogels with varied crosslinking rates in D2O were measured using a 60 MHz (Spinsolve 60) spectrometer. The water proton NMR relaxation times T2 was measured on hydrogels with different amounts of crosslinking. The T2-relaxometry was carried out using the Carr-Purcell-Meiboom-Gill (CPMG) pulse sequence and the experiments were performed at room temperature. The T2 was determined by fitting the echo amplitude decay according to the following equation:
FTIR-spectroscopic measurement
FTIR spectrometer was used to analyze various samples of PASP/GA hydrogel in order to verify the predicted cross-linking reaction, using Fourier-transformed infrared (FTIR) spectra (ATR-IR, Bruker, TENSOR 27). The powder sample was dried prior to this analysis.
Swelling ratio
Exactly 0.2 to 0.3 g of crushed hydrogel are weighed and inserted into two tea bags, which are then fastened together with bale ties. These bags are then placed in deionized water at 25°C, and the tea bags (minus the cable ties) are re-weighed at different time intervals (0.5, 1, 2, 3, 24 h).
The swelling rate is calculated using the following formula:
The Voigt model was utilized to calculate the maximum swelling ratio Smax and t63 (the time elapsed after 63% of Smax had been reached):
SEM observation
A scanning electron microscope (JSM-IT500HR) was used to analyze the surface morphology of Superabsorbent polymer PASP/GA after it had been swollen, followed by freeze-drying.
Results and discussion
The X-ray diffractometer results suggest that the copolymer is a random copolymer. The NMR spectra confirm the successful synthesis of the copolymer and its composition. FT-IR spectra reveal characteristic bands for the copolymer and the subsequent hydrogel.
Synthesis of PSI/GA1
The following part presents the formation of hydrogels from PASP/GA with the HMD, LYS or TD crosslinked. As for GA the optimum condition was obtained with the yield of 81% at the reaction temperature about 180°C, for 3 h, with the molar ratio of ASP acid to GA of 4.5:0.75 and catalyst concentration of 0.03 mol, this pre-polymer was used for hydrogel formation (Figure 3). Part (a) shows the reaction scheme for the preparation of PSI/GA, part (b) displays the chemical structure of the cross-links in PSI/GA, and part (c) details the cross-linking of PSI/GA and hydrolysis for the creation of PASP/GA hydrogels.
X-ray diffractometer
In Figure 4, the X-ray diffraction analysis demonstrates the absence of crystalline peaks. Instead, only two broad peaks are evident, corresponding to the amorphous phase of PSI/GA1. This finding suggests that both PSI and PSI/GA1 exhibit similar XRD patterns, with only slight variations in peak positions. As a result, it can be concluded that the introduction of glutamic acid (GA) into PSI did not substantially alter the amorphous nature of the material or its XRD pattern.19,21 X-ray spectra of PSI and PSI-co-glutamic acid.
NMR
Figure 5 displays the 1HNMR spectra of PSI and PSI/GA1. For PSI, signals at 2.7 and 3.21 ppm, and 5.27 ppm corresponded to the methylene and methine groups, respectively. Furthermore, the lack of any absorption in the region of 7.8-9ppm suggested the presence of branched and/or opened amide protons. PSI/GA’s 1HNMR spectrum featured signals at 2.7-3.21 and 5.27 ppm due to the methine protons of the succinimide units. Moreover, signals at 7.88 and 8.74 ppm, 4.91 ppm, 1.9-2.14, and 2.28 ppm were assigned to the amide, methine, and methylene protons of the glutamic acid units. Additionally, it is worth noting that GA introduced by 10% from the integration. 1HNMR spectra of PSI (a) and PSI/GA1 (b) in DMSO-d6.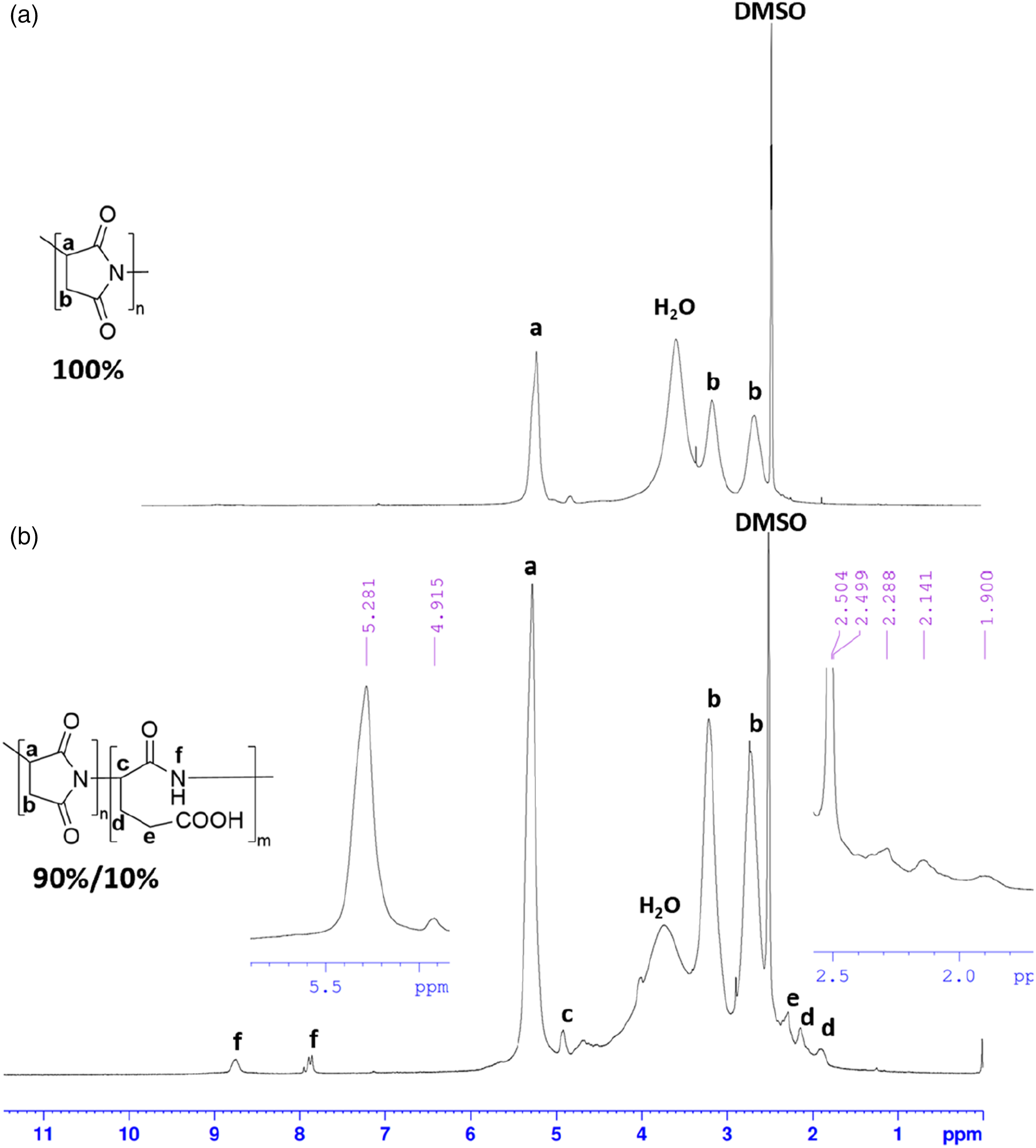
The 13C NMR Figure 6 of the copolymer showed four peaks at δ = 173.95, 172.67, 171.85, and 169.67 ppm associated with C = O, two peaks at δ = 47.93 and 52.20 ppm correlated to -CH-, and three peaks at δ = 32.81, 24.02, and 23.76 ppm connected to -CH2-, indicating the presence of both PSI and polyglutamic acid.19,20 13C NMR of PSI/GA in DMSO-d6.
Syntheses of tartaric acid dihydrazide (TD)
In addition to the HMD and LYS crosslinkers, tartaric acid dihydrazide (TD) crosslinker was prepared from tartaric acid using the method outlined above.
In the 1H-NMR spectrum of Tartaric Acid Dihydrazide (TD), two distinct signals were evident. The singlet at 4.54 ppm corresponds to the methine group (H-2, H-3), while another singlet at 4.70 ppm is attributed to both hydrazide protons and hydroxyl protons (Figure 7(a)). 1H-NMR spectra of tartaric acid dihydrazide (TD) (a) and 13C-NMR spectra of tartaric acid dihydrazide (TD) (b).
The 13C-NMR spectrum of Tartaric Acid Dihydrazide (TD) revealed distinctive peaks. A peak at 71.95 ppm indicates carbon atoms at positions 2 and 3, emphasizing their aliphatic nature. Another peak at 171.85 ppm aligns precisely with the carbonyl carbon of the amide group in the hydrazide moiety, confirming the presence of an amide linkage (Figure 7(b)).
The NMR spectra demonstrate the successful synthesis of the crosslinker, which was then employed in the subsequent reaction step to create PASP/GA-based hydrogels.
Characterization of PSI/GA, PASP/GA and the Chemical Structure of Hydrogels Using FT-IR Spectroscopy
In Figure 8(a), FTIR spectra of PSI, PSI/GA1 and PASP/GA are displayed. The spectra of PSI13,22 and PSI/GA present similar peaks, with the latter showing an additional absorption peak at 1352 cm−1 which is attributed to the -CONH group, indicating the presence of glutamic acid in the copolymer. Both spectra display characteristic bands at 1388 cm−1 (stretching vibration of -CONH groups), 1715 cm−1 (C = O) and 2952 cm−1 (stretching vibration of -CH2 groups).19–21 For PASP/GA, N–H groups (stretch corresponding to secondary amides) can be seen between 3250 cm−1 and 3300 cm−1, a peak at 1588 cm−1 corresponds to the -NH vibration from “amino acid”, and at 1392 cm-1 the -C = O vibrations of COO group are observed. The FT-IR spectra of clPASP/GA-HMD hydrogels created with varying quantities of 1,6-hexane diamine (crosslinker) are displayed in Figure 8(b). All samples contained a cross-linked CONH stretching band at 1518 cm−1. It was observed that when excessive amounts of cross-linking agent were used, the absorption peak at 1518 cm−1 was enhanced. Figure 8(c) of the FTIR results shows that the peak at 1518 cm−1 confirms the presence of –NH in the clPASP/GA-LYS 10.0, which is the end group of the cross-linkers. Finally, Figure 8(d) shows evidence that the Tartaric Acid Dihydrazide (TD) was successfully incorporated in the hydrogel structure due to the appearance of new bands at 1656 cm− 1 and 1518 cm− 1, which are stretching vibrations of the -CONH groups. FT-IR spectra of PASP/GA cross-linked with various cross-linking agents.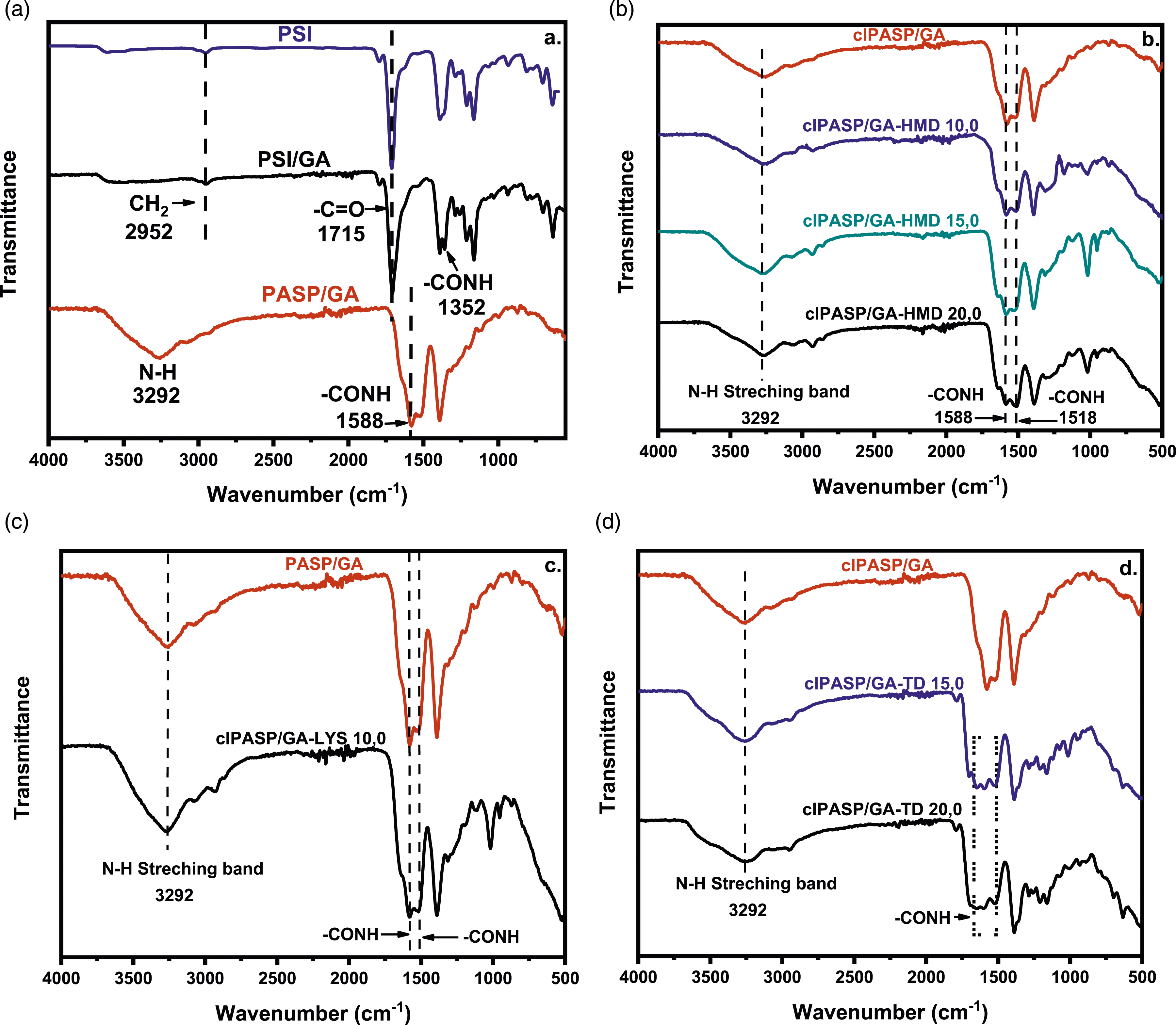
Influence of the crosslinking agent concentration on the swelling rate of PASP/GA hydrogels
The influence of the type and quantity of crosslinker can be seen clearly from the swelling ratio and kinetic parameters of the measurements. Figure 9 displays the kinetic data for the water absorption of various PASP-based hydrogels. The Smax and t63 parameters were calculated using the Voigt model, as shown Figure 10. Swelling kinetics of clPASP/GA-HMD, clPASP/GA-LYS and clPASP/GA-TD hydrogels. Dependency of experimentally determined maximum swelling degree on crosslinker content of (a) clPASP/GA-HMD, (b) clPASP/GA-LYS and (c) clPASP/GA-TD.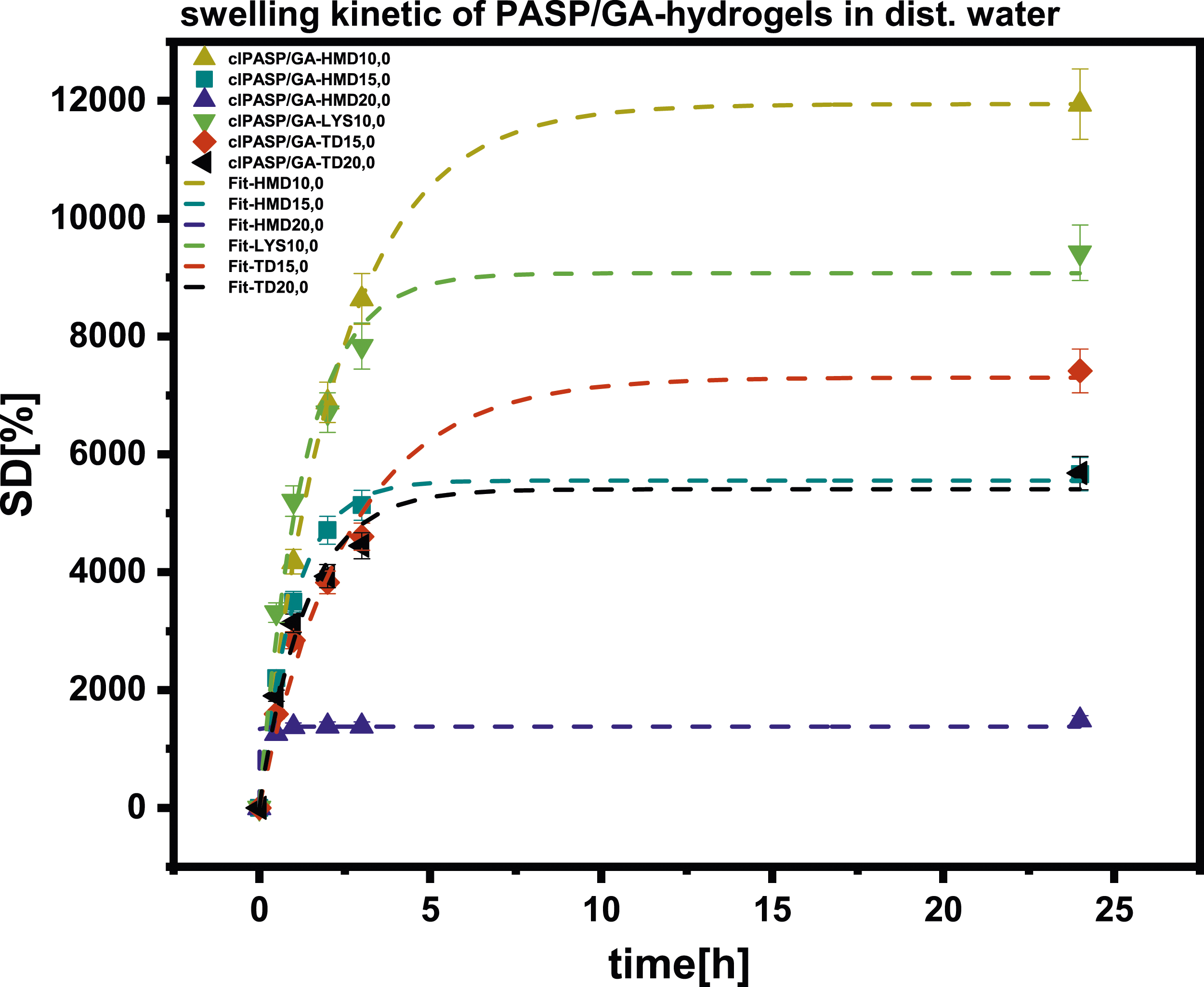

Swelling degrees between 1000% and 12000% were achieved when clPASP/GA was crosslinked with HMD, LYS, and TD respectively.
The addition of crosslinkers to hydrogels with HMD and TD demonstrated a decrease in the maximum water absorption (Smax) as the amount of crosslinker increased. This was accompanied by a decrease in t63, which indicated a slower uptake of the swelling medium. A decrease in water uptake with higher crosslinker concentrations is to be expected. In comparison to HMD, the incorporation of crosslinkers using TD was worse since no hydrogels could be formed at 10%. Unfortunately, there is insufficient data to provide information regarding 15% and 20% of the crosslinker (LYS). However, the swelling ratio of 9134% was achieved in the LYS hydrogel with 10%.
NMR measurements of T2 relaxation times for 0.5, 3, and 8 s
Figure 11 illustrates NMR measurements of T2 relaxation times performed on poly (aspartic-co-glutamic acid) (PASP/GA) hydrogels for durations of 0.5, 3 and 8 s, revealing a clear relationship between the degree of cross-linking and water mobility in the hydrogel Table 8. When the degree of cross-linking was increased from 10% to 20% in clPASP/GA-HMD samples, notable changes in T2 relaxation times occurred, indicating modifications in the mobility and behavior of water molecules within the hydrogel. The decrease in T2 relaxation times at the 20% degree of cross-linking implies a reduction in water mobility compared to lower degrees of cross-linking. Similarly, in clPASP/GA-TD samples, variations in T2 relaxation times were detected with increasing cross-linking from 15% to 20%. Specifically, T2 relaxation times decreased at higher cross-linking levels, signifying a decrease in water mobility in the hydrogel as cross-linking intensity increased.23–26 T2 relaxation times NMR measurements of PASP/GA cross-linked with various cross-linking agents for three different t: (a) 0.5 s, (b) 3 s and (c) 8 s. NMR measurements of T2 relaxation times (for three different times: 0.5, 3, and 8 s).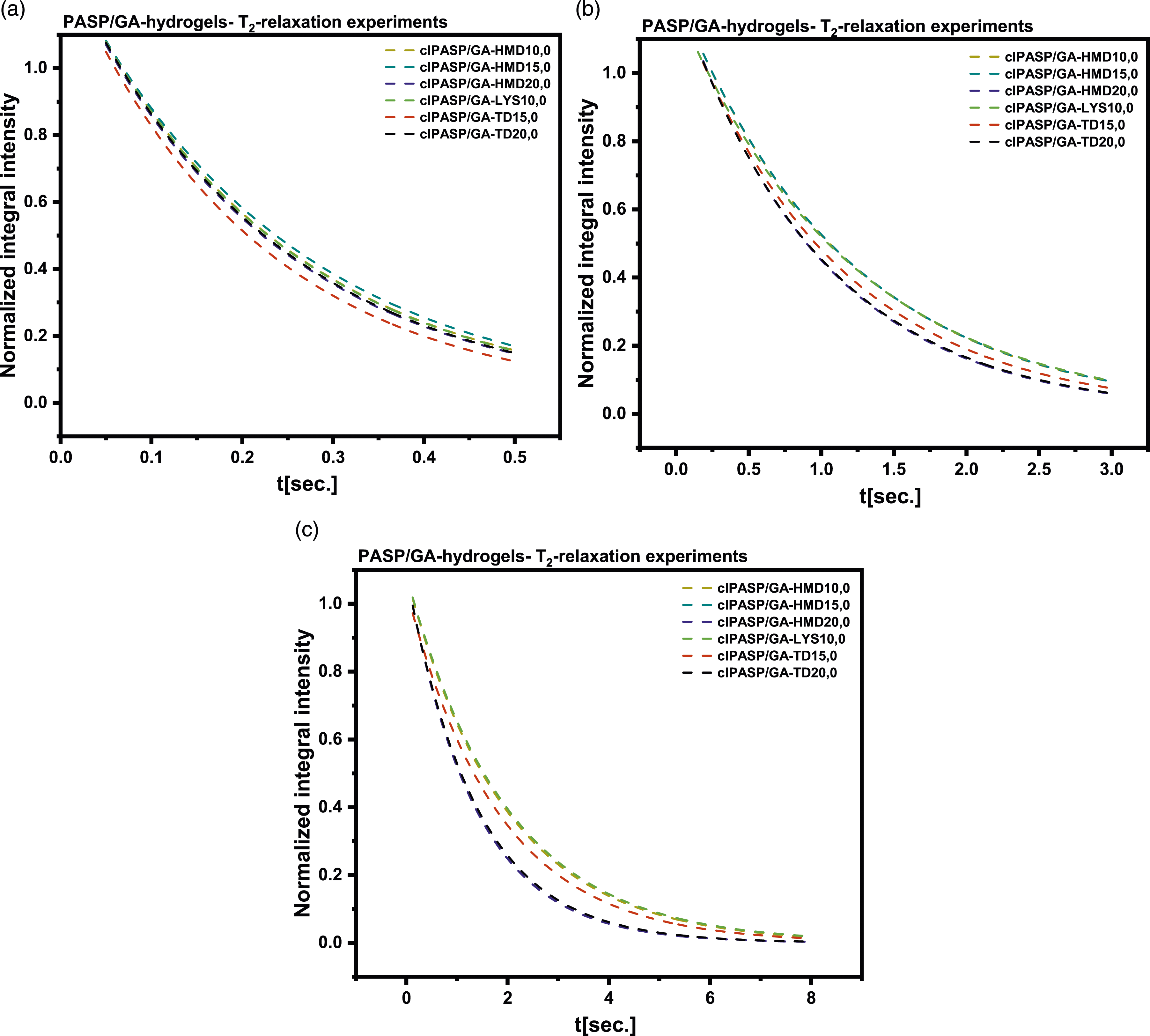

Morphological analysis
Figure 12 illustrates the morphology of hydrogels based on poly (aspartic-co-glutamic acid) with varying quantities of crosslinkers. The hydrogel exhibits a microporous structure, and as the proportion of hexamethylene diamine to poly (aspartic-co-glutamic acid) within the hydrogel rises, its pore size diminishes. This occurs because an excessive concentration of crosslinking agent results in an increased crosslinking density, consequently diminishing the hydrogel’s capacity to absorb water.27,28 Surface SEM images of (a) PASP/GA-HMD 10%, (b) PASP/GA-HMD 15% and (c) PASP/GA-HMD 20% hydrogels.
Conclusions
In conclusion, this study presents a biodegradable superabsorbent copolymer, merging D,L-aspartic acid (ASP) and L-glutamic acid (GA) through melt polymerization. Structural analyses confirm the copolymer’s unique physicochemical properties and efficient synthesis with yields reaching 81%. Strategic blending of ASP and GA capitalizes on their inherent advantages, resulting in a copolymer exhibiting remarkable water retention capabilities. The exploration of diamine cross-linking agents identifies optimal conditions, achieving a maximum swelling ratio of 11.874% with 10% hexamethylene diamine. Beyond contributing to copolymer synthesis and water absorption knowledge, this research stands out for its innovative combination of ASP and GA, offering a promising solution in the realm of sustainable superabsorbent materials. The copolymer’s potential applications underscore its significance, marking a noteworthy advancement in the pursuit of environmentally friendly alternatives to non-biodegradable polymers.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.