
Abstract
Polymers play an emerging role in modern engineering applications due to their comparatively low cost, low density, and versatile manufacturing. The addition of nano-sized fillers further enhances the polymer’s properties but also induces a strong dependence on the resulting microstructure, particularly the matrix-filler interphase. Since an experimental characterization of this nano-sized interphase is extremely difficult, molecular dynamics (MD) simulations are used to study the effects at such small scales. However, MD’s high computational costs usually limit the scope of a mechanical characterization. Therefore, this study presents the methodology and tools to generate and analyze samples of an efficient generic thermoplastic model. In this first contribution, we focus on the neat polymer and introduce a versatile and numerically stable self-avoiding random walker with adjustable linearity of chain growth. Moreover, we verify our equilibration procedure by preparing samples in liquid and solid state which behave physically sound. Finally, we perform uniaxial tensile tests with a maximum strain of 10 % to evaluate the mechanical properties. In the liquid case, the polymer chains are sufficiently mobile, such that the tensile stresses fluctuate only around zero, while the solid exhibits an almost linear elastic regime followed by a nonlinear part. This contribution forms the basis for a thorough mechanical characterization of polymer nanocomposites which we will address in future studies. The methodology and tools introduced are not limited to our generic polymer, but applicable to many coarse-grained models.
Introduction
Due to constantly growing demands on modern products, such as lightweight construction, environmental justice and cost optimization, the requirements on the materials used are likewise increasing. Polymers play an emerging role in this context since they are optimally adjustable to the application. They are versatile, cost-effective to process and offer enormous potential for use especially as nanocomposites. However, for a professional application, precise knowledge of the properties of a material and their influencing factors is essential.1,2
In engineering applications, the focus is particularly on the mechanical properties. The characteristic data and the material behavior are determined by various standardized tests which are time-consuming and expensive. Likewise, influences of the molecular microstructure, which are crucial for the fracture behavior and the outstanding properties of nanocomposites, cannot be determined by such tests. For this reason, simulations are increasingly performed, since they are more and more attractive due to steadily increasing computing power. They offer additional analysis capabilities, such as the possibility to explore the constitution of materials at much smaller length scales. The molecular level, which is particularly important for the mechanical behavior of polymers, is studied primarily with molecular dynamics (MD).
Molecular dynamics enables the mechanical characterization of the bulk properties as shown i.a. in Refs. 3–5, 6 and 7 for thermoplastics and thermosets, respectively. Furthermore, Odegard et al.8,9 derive continuum mechanical constitutive models directly from MD simulation with their equivalent continuum method. Additionally, MD reveals mechanisms that are hardly accessible experimentally, as, for example, the formation of an interphase region surrounding the filler particles in polymer nanocomposites.10–12 Even novel materials, such as ionic polymer nanocomposites, can be explored with molecular dynamics which was demonstrated by Moghimikheirabadi et al. 13 . However, the high computational cost associated with MD limits large scale studies, and thus more efficient models are desirable.
This work aims to outline the steps necessary to create virtual material samples of a Kremer-Grest model14,15 for a generic thermoplastic and to perform initial deformation simulations using a uniaxial tensile test. Different configurations of the test samples in liquid and solid form below the glass transition temperature are created and analyzed in terms of their stress-strain relation. In this first contribution, we demonstrate that the generic model at hand exhibits the characteristics of thermoplastic polymers. Hence, the introduced model is suited for future investigations, for example, on polymer nanocomposites.16,17 The subsequent characterization of the mechanical properties and the calibration of the appropriate constitutive laws will not be the aim of the work and are discussed in a follow-up contribution. 17
Methods
The mechanical investigation of virtual polymer samples requires the following four steps: Definition of the material properties, creation of virtual material samples, equilibration of the samples and the actual deformation simulation. Therefore, we present the material description of a generic, efficient thermoplastic in Subsection material description, introduce our new self-avoiding random walk algorithm in Subsection self-avoiding random walk algorithm, and describe the equilibration and deformation procedures in Subsections equilibration and deformation, respectively.
Material description
On an engineering-relevant scale, material models are usually described by continuum mechanics.18,19 As a consequence, the consideration of atomic properties and interactions are mostly not represented. However, these constitutive laws can be further improved by incorporating crucial molecular effects based on molecular dynamics simulations. Since the computational effort for these cannot consider sufficiently large particle systems in which surface influences can be neglected, periodic boundary conditions are used. In addition, for further reduction of computational cost, repeating atomic structures are substituted by so-called superatoms in the coarse graining (CG) approach. 20 This reduces the degrees of freedom, but also neglects subatomic effects. In general, coarse graining is divided into bottom-up and top-down strategies. 21 The former establishes a structural mapping based on atomistic reference simulations and thus transfers the chemical and physical characteristics to the coarse-grained scale. 20 Bottom-up coarse graining typically suffers limited transferability to different thermodynamic states 21 and distorted dynamics. 22 On the contrary, the coarse-grained potentials are calibrated based on experimental observations in the top-down approaches. The resulting models lack the chemical detail of bottom-up approaches but offer larger transferability and thus facilitate crucial insights into polymers’ structure-property relations.14,21
In general, the interactions of MD systems are based on potentials between the interacting particles while the overall thermodynamic state is defined by ensembles. 23
Unit system
We employ the Lennard-Jones reduced unit system 24 which is commonly used for studies on generic polymer models.25–27 Here, all quantities are given as multiples of fundamental mass m = 1, length σ = 1, energy ϵ = 1, and Boltzmann constant k B = 1. The conformity of the quantities with the fundamental time τ is maintained via ϵ = mσ2/τ. Consequently, all quantities presented in the following are unitless but can be mapped back to, for example, SI units, 28 which has been shown for commodity polymers.29,30
Interaction potentials



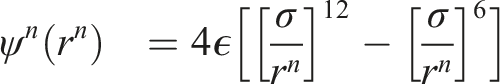
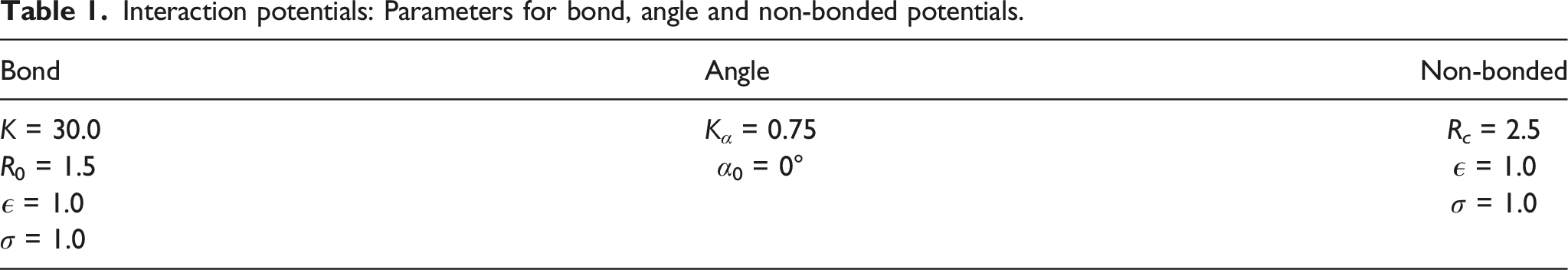
Interaction potentials: Parameters for bond, angle and non-bonded potentials.
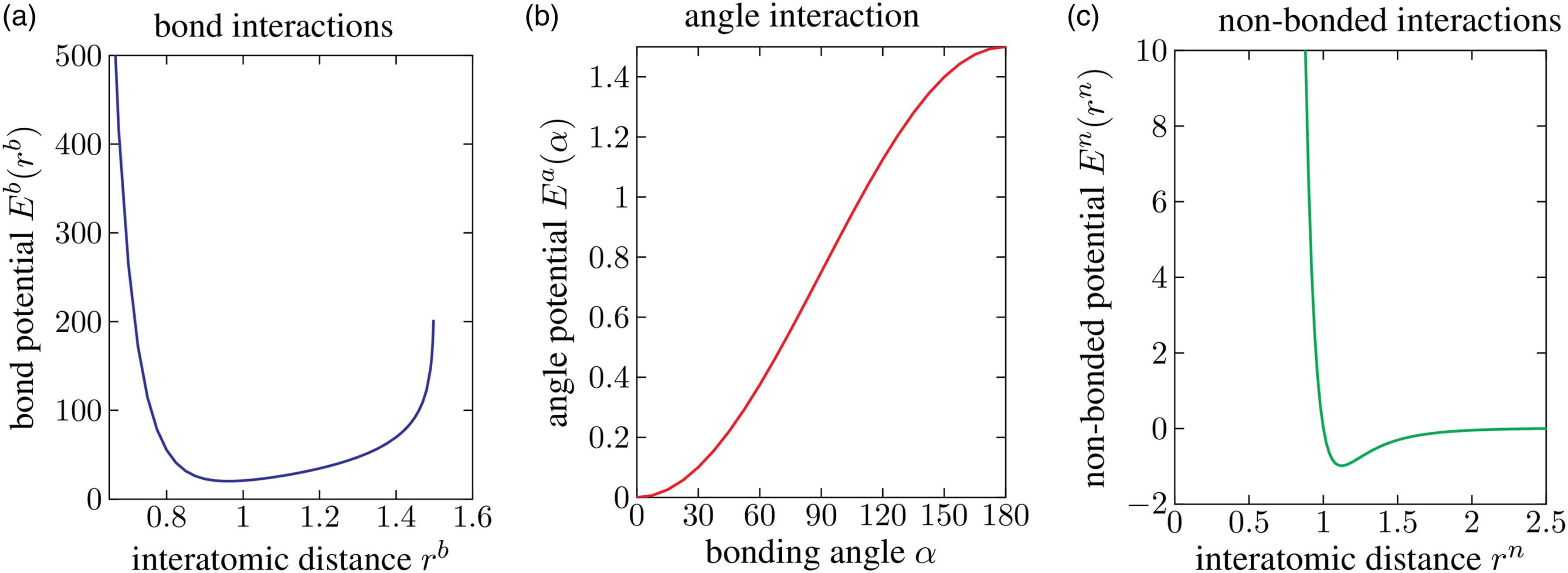
Visualization of the interaction potentials with parameters from Table 1: (a) bond, (b) angle, and (c) non-bonded potentials.
Self-avoiding random walk algorithm
After defining the particle interaction, we need to initialize their positions and connectivity to obtain the polymer samples. To this end, a self-avoiding random walk algorithm (SARW) 35 is introduced in the following.
The SARW’s objective is to generate a certain number of randomly arranged molecular chains with a defined number of atoms and a constant bond length. At least one atom per chain has to lie within the MD simulation box. Chains protruding from the box are taken into account via the periodic boundary conditions. The algorithm uses coarse graining and generates output files in LAMMPS’s molecular format. 32 Details on the random-walk procedure are given in the supplementary information Subsection S1 and the code is publicly available via Zenodo. 35
Input parameters of self-avoiding random walk algorithm.


Validation process for a new atom within the half opening angle θ and interatomic distance greater or equal to the bond length r
b
: (a) shows an invalid try for placing a new atom; (b) and (c) valid placement for a bead with a user-defined maximum allowed angle of θ = 120° for positioning new beads depicted in (c); In general, it holds for the accepted bond angle
Equilibration
After a material sample is created by means of the SARW, an equilibration is necessary. Hence, the interaction potentials from the material definition (cf. Subsection material description) are assigned to the created chains and a time integration is performed for a sufficient number of time steps to reach equilibrium. This means that the resulting sample converges to a stable state and is thus ready for deformation simulations. For this purpose, the thermodynamic state, that is volume, pressure, and temperature are controlled by means of the canonical (NVT) and the isothermal-isobaric ensemble (NPT).
Configuration ensembles and temperature sequences in time steps at constant pressure p = 0.

We initialize our samples at T = 1.0 since preliminary investigations have shown that the FENE bonds become numerically unstable at T > 1.0. Since this contribution is the first to evaluate the mechanical behavior of the CG model introduced in Subsection material description, only the lower (T → 0.0) and upper (T = 1.0) limit cases are considered. Note that the glass transition temperature of the present material was identified at T
g
≈ 0.41 in a subsequent study.
17
Since absolute zero temperature cannot be reached due to the employed Nosé-Hoover thermostats and barostats, we apply
Deformation
Various deformation simulations can now be carried out with the obtained samples. For a comprehensive analysis of the mechanical properties and the subsequent calibration of material laws, time-proportional, time-periodic as well as relaxation tests are required. 4 These simulations allow for a classification into elastic, plastic, viscous, viscoelastic or viscoplastic material behavior.38,39
However, this work aims at verifying the SARW and establishing an equilibration procedure in order to provide the basis for future investigations. Thus, we only consider the uniaxial tensile test and apply a maximum strain of 10.0 % within 
Results and discussion
This section discusses the results of the equilibration procedure and the uniaxial tensile tests introduced above, which are applied to the generic polymer model used throughout this manuscript. Since we observe a tangle-like chain growth with few entanglements between multiple chains in the SARW (cf. Figure 3(a)), we examine the impact of the initial material configuration during the equilibration and uniaxial tensile test by extending the SARW in Subsection influence on the bond angles and linearity of chain growth with a partially adjustable bond angle and linearity of chain growth to perform a short parameter survey. Self-avoiding random walk algorithm with four different, examplary angle configurations: (a) θ = 120°, (b) θ = 90°, (c) θ = 50°, and (d) θ = 5°.
Influence on the bond angles and linearity of chain growth
In order to get even better control of the material generation, we impose certain constraints on the step-wise selection of new atoms without completely excluding randomness. The individual chains tend to grow like balls and thus form only few entanglements with other polymer chains, as discussed in detail in Subsection equilibration at different temperatures. To avoid this and particularly to allow for more entanglements among the polymer chains, we include an adjustable linearity of the chain growth into the code, cf. Figure 3.
In this context, we would like to remark that this also increases the numerical stability together with the flexibility of the SARW. For this purpose, the chosen option specifies an allowable angle at which the new atom may be placed. Attempts to place an atom outside the angle are not accepted and new tries are started. Furthermore, the permitted distance to other atoms has to be fulfilled, which is why the condition is automatically relaxed the more often no permissible position is found, cf. Figure 2.
The mathematical relations for implementing this procedure in the SARW can be obtained from Subsection S2 and the code is publicly available via Zenodo. 35 As shown in Figure 3, constraining the half opening angle θ causes the chains to exhibit different degrees of linearity in chain growth and thus different initial bond angles ϕ, depending on the angle θ. 1 In general, choosing large maximum angles θ triggers a ball-like chain growth as discussed above and it has turned out that this makes the algorithm more likely to fail. Possible remedies are to increase the simulation box size, to decrease the maximum angle θ, or a combination of both.
For the short parameter survey, we include a broad range with four different angular configurations by setting θ to 35°, 50°, 60°, and 110°, respectively.
Equilibration at different temperatures
Two different target temperatures are evaluated for the equilibration. The target temperature of T = 1.0 will create liquid material samples with low viscosity (no cooling during equilibration, cf. Table 3), while T → 0.0 (numerically applied as Density during equilibration in NPT and NVT ensemble for the four samples with different initial angle constraint θ: (a) without cooling process for maintaining the target temperature T = 1.0; (b) with cooling process to target temperature T → 0.0.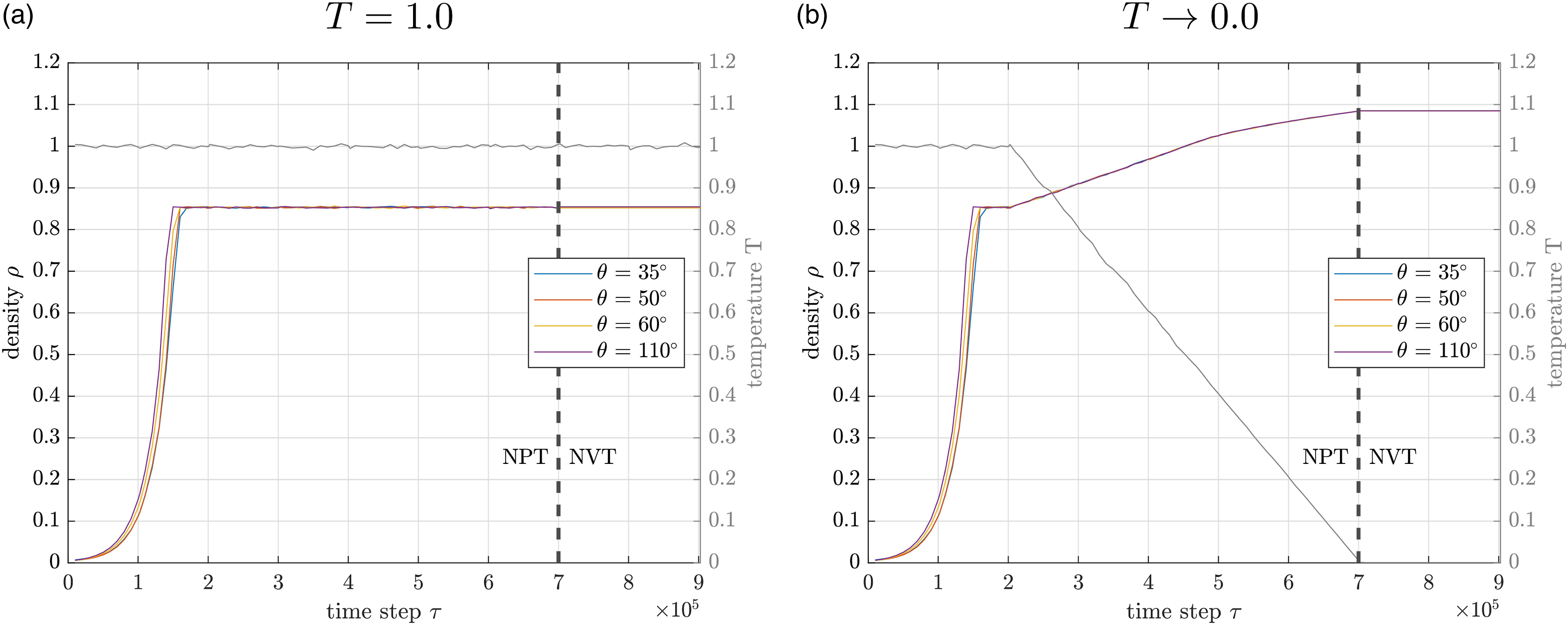
After the energy minimization, an exponential growth of density takes place over nearly
A difference between the four different angular configurations from the SARW is only apparent over the first
Since the SARW generates relatively large gaps between the individual chains, the selected potentials reduce the distances between them and change the bond angles ϕ accordingly. Thermostat and barostat have been checked and are regulating the system as specified. Furthermore, the increase in density after
To explain the small differences between the chains with different angular configuration, the distribution of bond angles between three atoms each is studied before and after equilibration for T → 0.0 in Figure 5. Note that most of the bond angles ϕ addressed here are Distribution of bond angles for the four samples with different initial angle constraint θ (cf. subsection 3.1): (a) before and (b) after equilibration of the samples at T → 0.0. The small sketch in (a) visualizes the increased probability that a random unconstrained atom has significantly more spherical surface available for possible positions in the range of a bond angle of 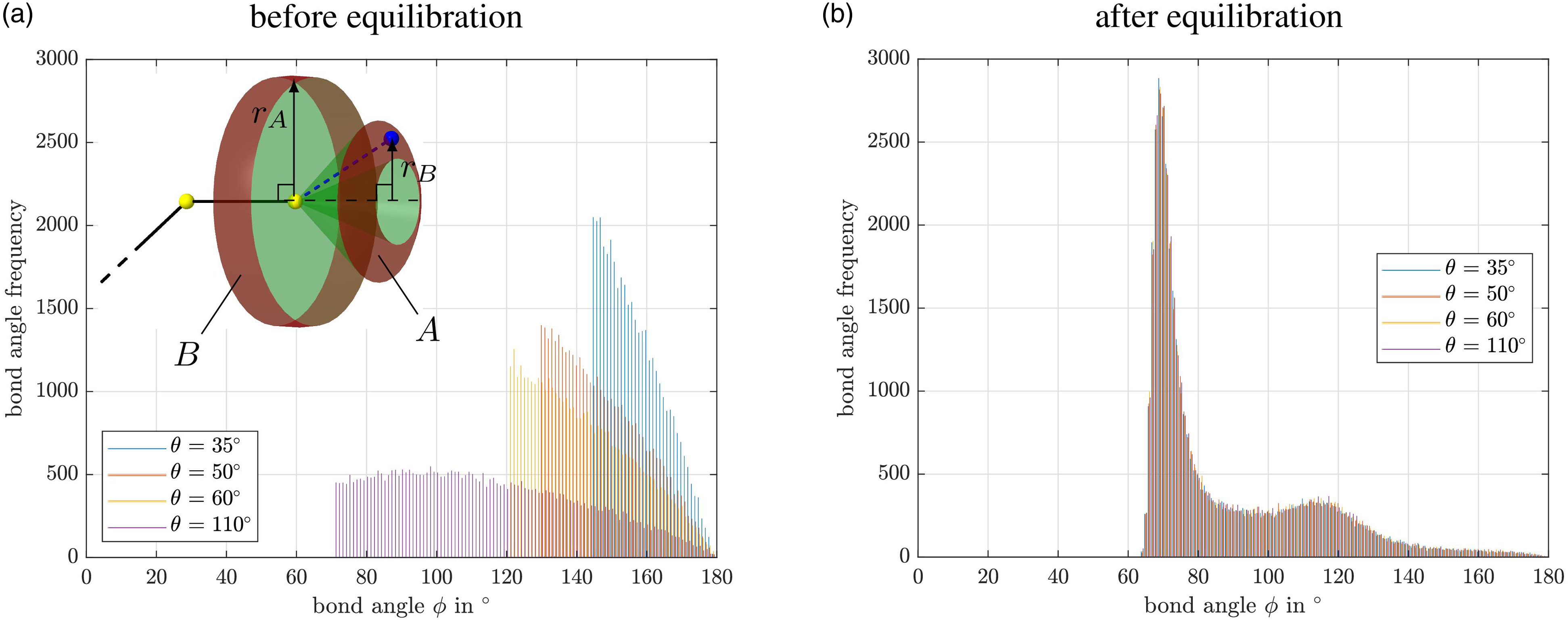
Before the equilibration, clear systematic differences are present in the individual samples as depicted by the histograms in Figure 5(a)). There, the frequencies of bond angles are given for different user-defined half-opening angles θ of the SARW. Due to the mentioned relation between θ and the initial bond angle ϕ, the different values of θ lead to different minimal possible bond angels for the four samples. The larger θ is chosen, the smaller the minimum bond angle ϕ gets. As expected, no angles smaller than
Such a behavior is based on the increased probability that a random unconstrained atom has significantly more spherical surface available for possible positions in the range of θ ≈ 90° than at a smaller θ. For example, it is shown in Figure 5 that a sphere surface segment of area A at bond angle 155° has only
After the equilibration, however, no differences between the individual material samples are visible anymore, cf. Figure 5(b). All angles are larger than 60°. At about 63°, the bond angle frequency increases very strongly until it reaches a maximum at
Markedly, Figure 5 shows that the influence of the half opening angle θ vanishes during the equilibration. After the simulation, all material samples have the same bond angles on average. It was expected that no angle smaller than 60° would be present in the materials, since the first and third atoms are so close due to the non-bonded potential that large repulsive forces are generated, preventing smaller angles. The most favorable configuration for the angles appears to be only slightly larger. There, most angles reach their equilibrium, which is a direct consequence of the defined interaction potentials. Due to the polymer’s amorphous microstructure, less favorable configurations exist as well, for example caused by entanglements. In these cases, bonded and non-bonded pair interactions are more dominant, enforcing wider angles resulting in the second local maximum at
In conclusion, the equilibration procedure discussed here, together with the SARW introduced in this contribution, allows for a reliable and efficient creation of material samples, which can also be used for other coarse-grained materials. As demonstrated in Figure 5, the chosen parameters of the SARW do not affect the final structural properties of the material for the generic model introduced in Subsection material description. Thus, our numerically robust equilibration procedure, which is in fact insensitive to the artificially chosen chain linearity, is well suited to obtain virtual material specimens for subsequent deformation simulations.
Furthermore, the density evolutions in Figure 4 provide an estimation for the number of time steps necessary for the samples to converge. Since the equilibration covers the complete temperature range of a solid sample, it is possible to derive the necessary number of time steps at other target temperatures.
Uniaxial tension
We apply uniaxial deformation, as discussed in Subsection deformation, in x-direction and evaluate the stress response σ
xx
for T = 1.0 and T → 0.0 in Figure 6(a) and (b), respectively. For the entire simulation at T = 1.0, the graphical representation shows fluctuations of the stress curve around the zero value for all material samples. The variation range is Stress-strain diagram of the uniaxial tensile test with up to 10 % strain in NPT ensemble for the four samples with different initial angle constraint θ: (a) samples with T = 1.0 fluctuate around σ
xx
≈ 0; (b) samples with T → 0.0 have a linear increase in σ
xx
first before the stress-strain relation gets nonlinear and reaches a plateau.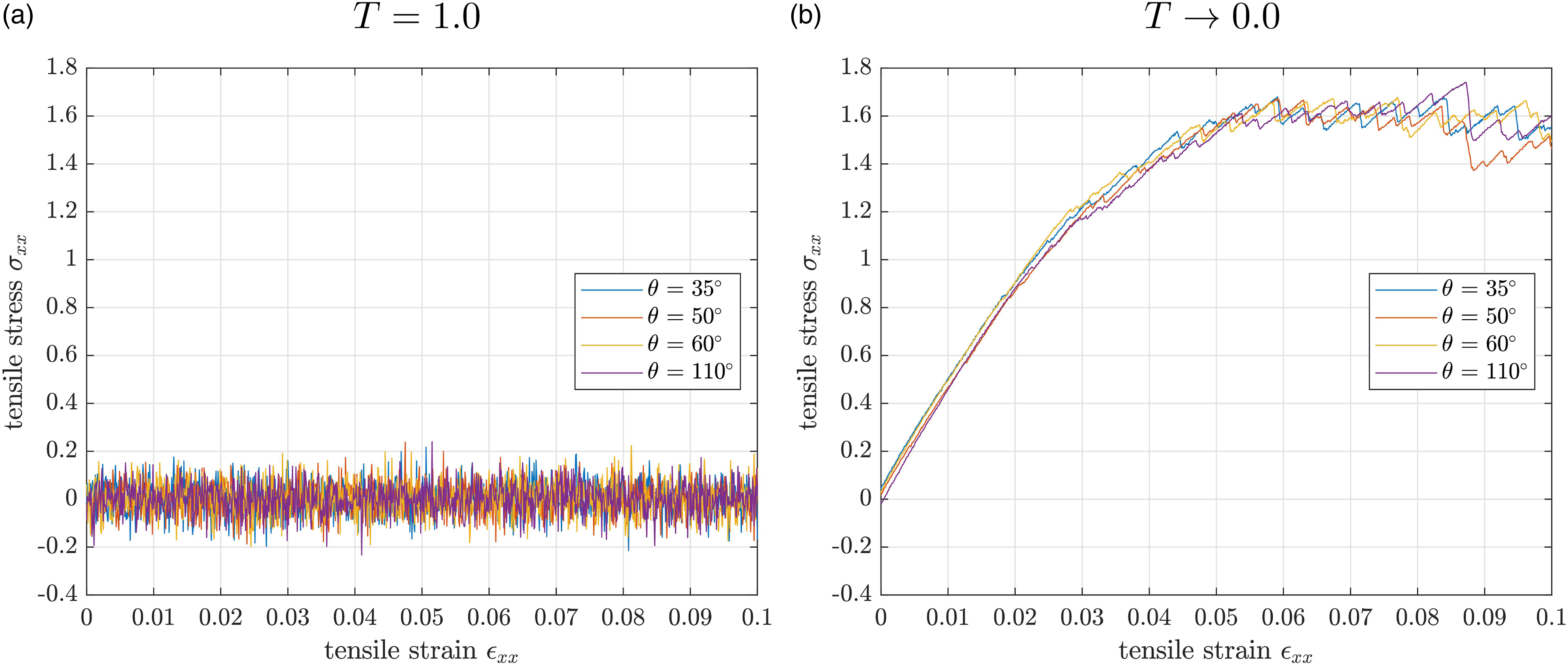
For both temperatures the stress behaves as expected: The viscosity of the liquid material samples (T = 1.0) is low enough for an instantaneous and full stress relaxation, even when 10% strain is applied. The fluctuating behavior is attributed to the temperature-induced high mobility of chains.
The samples at T → 0.0 and thus below the glass transition temperature, reveal the typical stress-strain behavior of solid polymers.
42
In the linear elastic region, the Young’s modulus
Determining Poisson’s ratio for a liquid material is not possible due to the strong fluctuations. For the solid at T → 0.0, the lateral strain in y-direction ɛ
yy
is plotted over the tensile strain in x-direction ɛ
xx
in Figure 7. Plot of strain in y-direction over strain in x-direction for the samples at T → 0.0.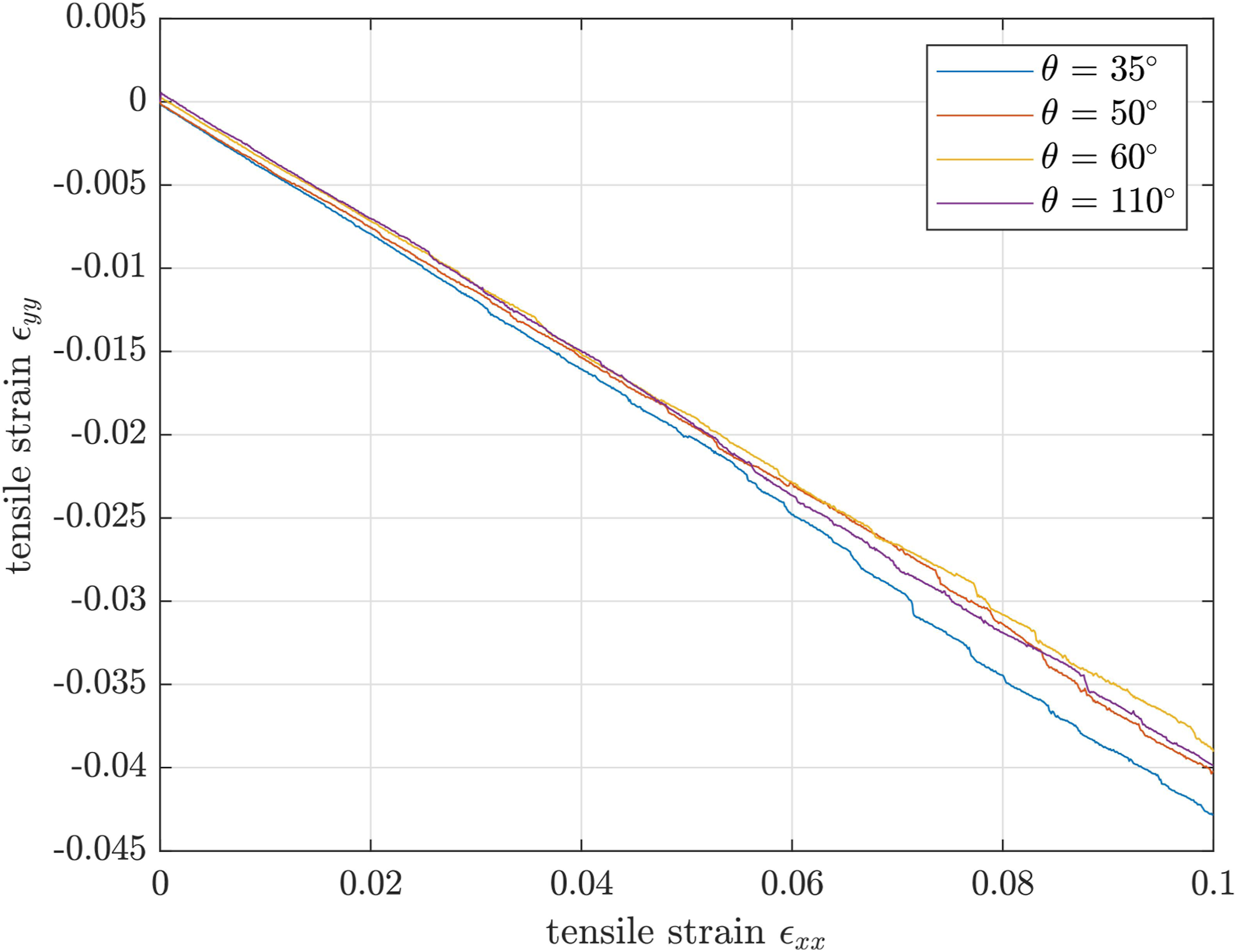
There is nearly no difference in the behavior of the individual samples and an almost completely linear curve is evident which renders an average Poisson’s ratio
In addition to the conventional diagrams used to characterize mechanical behavior, the spatial arrangement of the chains is analyzed. Therefore, the movement of a single chain for a sample with T = 1.0 and T → 0 is observed during the uniaxial tensile test in Figure 8. Investigation of the spatial arrangement for an isolated chain every 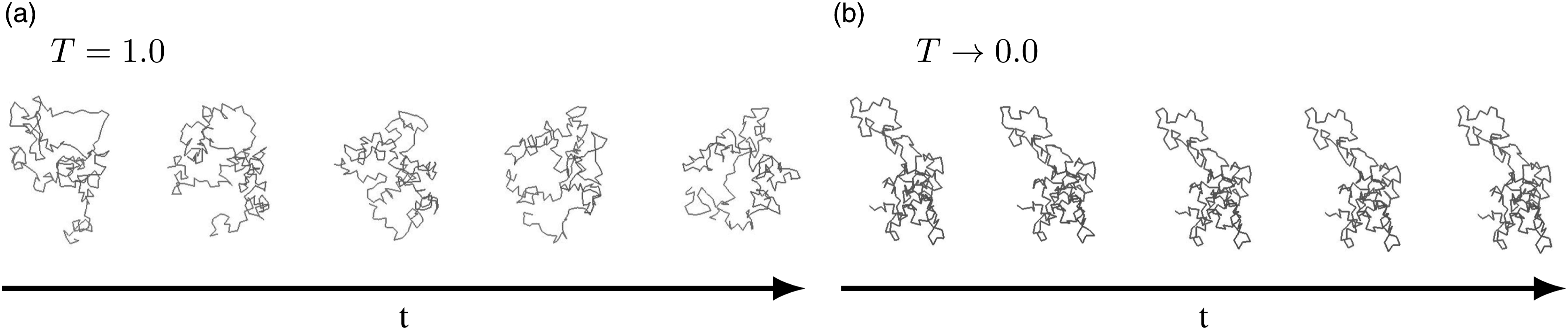
It is clearly visible that within the considered time span in the tensile test at T = 1.0 the chain structure as well its local position change considerably. Stresses can thus be relieved due to chain mobility. In contrast, very little topological change is evident in the samples below the glass transition temperature, cf. Figure 8(b)).
The equilibration and deformation using the uniaxial tensile test at different temperatures and bond angle distributions as shown here can be used as a reference for similar systems. A complete mechanical characterization exceeds the scope of this paper and can be obtained from the follow-up contribution. 17
Conclusion and outlook
In this work, the four essential steps to create virtual material samples for a generic polymer model have been thoroughly discussed. First, the properties of the material were defined using appropriate potentials. Then, an efficient, stable algorithm for the controlled creation of virtual samples with adjustable linearity of chain growth was presented. Different input parameters allow for a wide range of individual settings and thus applicability to many coarse-grained models. For the third step, the equilibration, different configurations were generated and the limited cases of T = 1.0 (liquid state) and T → 0.0 (solid state) have been studied. As a result of the SARW algorithm, different distributions of bond angles occur before equilibration, which, however, do not affect the resulting polymer systems after equilibration. In the simulations, the process for reaching an equilibrium state at the boundary temperatures of T = 1.0 (liquid state) and T → 0.0 (below glass transition temperature) was performed and analyzed. The results show that the samples behave physically sound, that is a liquid with low viscosity and comparably low density ρ ≈ 0.85 is obtained for T = 1.0, whereas a solid sample with higher density ρ ≈ 1.09 is achieved in case of T → 0.0. Furthermore, tensile tests with a maximum strain of 10 % have been performed, which also yield plausible results: In the liquid case, the polymer chains are sufficiently mobile, such that the tensile stresses fluctuate only around σ xx = 0. In contrast, at low temperature, the stress-strain diagram exhibits an almost linear elastic part with a Young’s modulus of E ≈ 41.5, followed by a nonlinear part with a yield stress of σ s ≈ 1.62. Furthermore, a Poisson’s ratio of 0.375 is obtained in this case.
In summary, the procedure shown here for a generic polymer model can be used as a basis for a wide variety of future studies. In this regard, the self-avoiding random walker publicly available via Zenodo 35 provides a practical and reliable mean to generate numerical representations of thermoplastics at coarse-grained resolution, which form the basis for our subsequent studies: As a first step, we are performing a thorough mechanical characterization of the generic polymer. 17 As a second step, we incorporate nano-sized filler particles into the generic polymer to enable a mechanical analysis of polymer nanocomposites 17 and their matrix-filler interphase. 16
Supplemental Material
Supplemental Material - Studying the mechanical behavior of a generic thermoplastic by means of a fast coarse-grained molecular dynamics model
Supplemental Material for Studying the mechanical behavior of a generic thermoplastic by means of a fast coarse-grained molecular dynamics model by Vincent Dötschel, Sebastian Pfaller and Maximilian Ries in Journal of Polymers and Polymer Composites.
Footnotes
Acknowledgements
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Sebastian Pfaller is funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) - 396414850 (Individual Research Grant ’Identifikation von Interphaseneigenschaften in Nanokompositen’). Maximilian Ries and Sebastian Pfaller are funded by the DFG - 377472739 (Research Training Group GRK 2423 ’Fracture across Scales - FRASCAL’).
Simulation software and data
The MD calculations were performed with LAMMPS (version 29 Oct 2020), 46 while the parameter identification of the continuum mechanical models was done in Matlab.
Supplemental Material
Supplemental material for this article is available online.
Note
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.