
Abstract
Uniform and nearly monodisperse superparamagnetic Fe3O4/Poly(methyl methacrylate) (core)/SiO2 (shell) nanoparticles with raspberry-like morphology and high saturation magnetization were prepared in three different steps. At first, a facile, one-shot procedure to synthesize hydrophobic Fe3O4 nanoparticles through a modified co-precipitation method was implemented. Based on the hydrophobic interactions, these nanoparticles were used directly in a mini-emulsion polymerization resulting in encapsulation with PMMA. Then, for the covering with a silica shell, the surfaces of the Fe3O4/PMMA nanospheres were hydrolyzed in alkaline media and became hydrophilic through hydrolyzation. In the last step shell deposition of the Fe3O4/PMMA nanospheres through a modified Stober method was implemented. The surface morphology was investigated by scanning electron microscopy (SEM) and the core-shell structure and the prepared products’ diameters were measured by transmission electron microscopy (TEM); the size of the magnetic nanospheres was approximately 83 nm. Vibrating sample magnetometry (VSM) showed high magnetic saturation and superparamagnetic characteristics of the particles. Thermogravimetric analysis (TGA) was used as a supplementary test and, based on the mass loss at high temperature (600°C), the magnetic (Fe3O4) and non-magnetic content (PMMA) of the Fe3O4/PMMA nanospheres was measured as 81 and 19%, respectively. The narrow polydispersity of the nanospheres, measured by dynamic light scattering (DLS), was approximately 0.101. In every preparation step, the synthesized products were characterized by Fourier transform infrared (FTIR) spectroscopy. Our study focused on designing two-layered magnetic nanoparticles with drug delivery potential using two-layer encapsulation based on the hydrophobic and hydrophilic surface characteristics of the PMMA core and silica shells, respectively.
Keywords
Introduction
Recent developments in the field of biotechnology have introduced inorganic and organic nanocomposites owing to their intriguing properties.1–6 Among a wide range of nanocomposites, magnetic polymer composite nanospheres have profound merits in many biomedicine applications because they can be easily collected under a magnetic field. The promising applications of these MNPs (magnetic nanoparticles) involve drug delivery,1,2 magnetic resonance imaging (MRI),
3
hyperthermia,
3
nucleic acid detection,
4
enzyme immobilization,
5
magnetically assistance site-specific drug delivery,
6
and water treatment.
7
Magnetic polymer particles in the field of biomedicine should possess some exclusivity, such as a narrow nano-sized distribution, high and uniform magnetic content, and high saturation magnetization (MS),
8
no sedimentation,
9
and nontoxicity.
10
In the use of MNPs in cancer treatment, there are three specific properties of concern: 1. The size of the NPs 2. A high surface area 3. A functionalized surface in order to enhance targeting cancer drugs.
11
The naked MNPs aggregate due to their magnetic dipolar attractions; therefore, they need to be encapsulated for biomolecule immobilization with high stability12–14 but remain magnetic. As surface engineering of the MNPs is a particular issue for encapsulation with polymers, as we showed previously, they should possess a compatible surface with the polymer shell which emphasizes the need for surface modification of the MNPs. 15
Diverse approaches have been applied for the preparation of magnetic/polymer composite particles. To our knowledge, Guesdom and Avrameas first synthesized magnetic polymer spheres, using acrylamide and agarose in the presence of iron oxide NPs.
16
Encapsulation of MNPs can be conducted by various polymerization methods, such as emulsion polymerization, soapless emulsion polymerization, inverse emulsion polymerization, inverse microemulsion polymerization, seed precipitation polymerization, and so on. All these approaches produce various shapes (nanoflowers, nanospheres, nanorods, nanocubes, etc.) in three kinds of nanoparticles
11
: P/M (polymer particle core/magnetic nanoparticles shell),
17
P/M/P (polymer particle core/magnetic shell/polymer shell),
18
and MP (magnetic core/polymer shell
19
or magnetic NPs dispersed in a polymer matrix
20
). Recent studies have demonstrated the usefulness of suspension polymerization to synthesize core-shell NPs; Chen et al., for instance, applied suspension polymerization to synthesize PMMA/Fe3O4 microspheres.
21
Still, their produced microspheres suffered from irregular morphology, rough surface, and weak uniformity due to the addition of the Fe3O4. Other studies have been implemented to investigate the mini-emulsion approach for nanoscale particles. Yan et al. prepared Fe3O4/polystyrene composite particles via mini-emulsion polymerization,
22
but the polydispersity of the MNPs increased with the addition of more Fe3O4 nanoparticles which had been modified with oleic acid. Ma et al. conducted a new mini-emulsion polymerization approach to synthesize Fe3O4 NMPs encapsulated with poly(methyl methacrylate-acrylic acid).
23
In their research, NMPs with good hydrophilicity and high saturation magnetization were synthesized. Mahdavian et al. prepared Fe3O4-poly(butyl acrylate-styrene) via initiator free mini-emulsion polymerization; in their research, the Fe3O4 NMPs was located in the core and poly(butyl acrylate-styrene) encapsulated the magnetic core
24
and the core-shells were obtained under ultrasonic irradiation. The nanospheres were uniform, but the saturation magnetism was reduced sharply due to various factors, such as the addition of OA as a surface modifier, ultrasonic use (which can oxidize the Fe3O4 to
By considering the drawbacks of the various polymerization methods, mini-emulsion polymerization is the most efficient way because, in this method, the magnetic nanoparticles in the mini-emulsion media locate in the monomer droplets, and the polymer chains can encapsulate the nanoparticles. 26 For the preparation of Fe3O4/PMMA composite nanospheres, there has been extensive research. Lan et al. prepared nearly monodispersed Fe3O4/PMMA composite nanospheres with an MS content of about 39 (emu.g−1). 8 Another article by these authors used the same preparation method for superparamagnetic composite Fe3O4/PMMA nanospheres with 10–90 nm size for application in protein separation. 27 They synthesized another shape of these NPs with this approach; the superparamagnetic SiO2/(PMMA/Fe3O4) particles were prepared with a modified mini-emulsion polymerization and then coated with a SiO2 layer via electrostatic interaction, 28 resulting in the outer silica shell being periodically mesoporous. There are also some reports about implementing two-layer shells. Wang et al., for instance, prepared magnetic nanospheres (PMMA/Fe3O4) coated by a silica shell by the Stober process with separation and DNA enrichment applications. 29 He et al. reported the surface modification of Fe3O4 nanoparticles in the SiO2/(PMMA/Fe3O4) core-shell nanostructures; 9 but, after the surface modification, the magnetic saturation decreased notably, probably due to the thick shell, and the particles were not monodisperse in size. Another research report described the use of linolenic acid as a crosslinking agent for nucleic acid detection by the chemiluminescent method. 30 In this approach, the size distribution range was 250–500 nm, all with the same shape, and high specificity, and good sensitivity properties.
Herein we report the synthesis of superparamagnetic PMMA (as core)/SiO2 (as shell) nanoparticles, which have attached Fe3O4 nanoparticles resulting in a raspberry-like morphology, by mini-emulsion polymerization and the Stober method. Before the mini-emulsion process, the Fe3O4 NPs were hydrophilic and needed to be modified. NPs which contain hydroxylated surfaces of metal ions (Fe3O4, Al2O3, and TiO2) can be functionalized with carboxylic agents.21,31,32 Besides hydroxyl groups, magnetic metal-based NPs with hydrophilic surfaces may also contain surface-metal ions. This surface can be functionalized with biofunctionalized ligands, where one functional group is metal-coordinating and can be grafted on the surface of the metals. Simultaneously, the remaining chain has a weaker affinity for metal coordination and remains free for further reactions. 33 Until now, many surface modifiers for Fe3O4 have been applied. 29 Oleic acid, for instance, can make Fe3O4 dispersible in nonpolar media and can control particle agglomeration. Here, we introduce a modified co-precipitation method to synthesize hydrophobic NPs of Fe3O4 directly. In this method, the aggregation of NPs in the core of the nanostructure can be prevented effectively by the carboxylic acid group hindrance of oleic acid. In earlier studies by Lee et al., 30 an MMA’s polymerization process for encapsulation of Fe3O4 NMPs modified with OA produced many PMMA microspheres without magnetic NPs, and some of these modified NPs could agglomerate easily. For overcoming these consequences, we introduced OA during the Fe3O4 co-precipitation method. The next step was a mini-emulsion polymerization. The hydrophobic Fe3O4, MMA, and surfactants were sonicated and formed a mixed mini-emulsion with a homogeneous size dispersion of monomer droplets containing NMPs in water before the polymerization initiation. Modification with a polymer layer can preserve the colloidal stability through steric hindrance and increase the critical concentration of Fe3O4 MNPs during the next step. 29 The last step was silica coating with high drug delivery capacity. 34 Another benefit of this layer was the silane agents' reaction, which produced a stable dispersion in non-aqueous media. The silica coating was applied via the in-situ Stober method after hydrolysis of the nanospheres35–38 and the final product had an excellent spherical shape with superb monodispersity.
Materials and methods
Materials
Ferric chloride (FeCl3, 6H2O, Merck Co., USA) and ferrous chloride (FeCl2, 4H2O, Merck Co., USA) were used as received. Methyl methacrylate (MMA, Sigma-Aldrich Co., Ltd., Germany) and sodium dodecyl sulfate (SDS), citric acid (CA), and tetraethoxysilane (TEOS) all from Merck Co. were all analytical grades. Oleic acid (OA, 90% Merck) was used without purification. In each step, doubly distilled and deoxygenated (DI) water was used.
Characterization
The hydrodynamic diameter and size distribution of both the Fe3O4 and (Fe3O4/PMMA)-SiO2 MNPs were characterized by dynamic light scattering (DLS, model BI-200SM, Brookhaven Instrument Co., USA, with the wavelength of 657 nm). The Fourier transform infrared (FTIR) spectroscopy, by a spectrometer (MB Series, Bomem Inc., Canada) was used for investigating the functional groups with KBr discs. The mass loss of MNPs was obtained through thermogravimetric analysis (TGA, Pyris 1 TGA, PerkinElmer Corp., USA). The mass loss measurements of the dried MNPs were carried out under N2 with a heating rate of 10°C.min−1 from 35 to 700°C. Scanning electron microscopy (SEM) imaging of the surfaces of the MNPs was carried out by using an AIS2300 C SEM device (Seron Technology Co., Korea). The dilute aqueous solution of the sample was sonicated for 15 min by a Misonix sonicator (S3000, Misonix Inc, USA), then multiple drops of the sample were dropped onto a small aluminum foil and it was dried at room temperature. Then, the specimen surface was coated with gold (gold thickness around 10 nm). The sample was observed by the SEM. The internal structure and core-shell morphology were observed by transmission electron microscopy (TEM, EM 900, Zeiss Co., Germany) operating at 100 kV. The dilute aqueous solution of the sample was also sonicated for 15 min by a Misonix sonicator (S3000, Misonix Inc, USA), then one drop of the sample was dropped onto a formvar carbon film on copper grid 300 mesh (EMS-USA) and it was dried thoroughly at room temperature. The sample was observed by the TEM. The magnetization measurements of the samples were obtained by a vibration sample magnetometer (VSM, Meghnatis Daghigh Kavir Co., Iran) with a field from 0 to 10,000 Oe at 300°K. Al samples were measured three times, and the average values were reported.
Preparation of Fe3O4 nanoparticles modified with OA
The MNPs were produced via a modified chemical co-precipitation method with the preparation being implemented under N2. Two aqueous solutions, 75 mL containing 0.023 mol FeCl2 and 75 mL containing 0.046 mol FeCl3, were prepared separately. The molar ratio of FeCl2/FeCl3 was 1:2, based on Massart’s method for the preparation of Fe3O4 NPs. 39 The two aqueous solutions were mixed under N2 and ultrasonic treatment (50 W, 2 cycles with 2 min duration) and then transferred to a 250 mL four-necked-flask equipped with a condenser, nitrogen inlet and a mechanical stirrer (400 r/min). Then, 50 mL of ammonium hydroxide (NH3OH, 25%) was added in drops (fractionally) under vigorous stirring. Following the NH3OH addition, after 30 min of stirring at 60°C, 2 mL of OA was added drop by drop under vigorous stirring (at 400 r/min), and then the flask contents were stirred for 1 h at 70°C. The resulting black oily product was collected on the flask wall by a magnet and washed 2 times with ethanol and 3 times with DI water in the presence of an external magnetic field, then dried in an oven at 60°C for 12 h. The washing procedure in this stage has an important effect on the next stages, and it was continued, as above until the pH of the remaining water reached 7.
Preparation of Fe3O4/PMMA nanoparticles
Fe3O4/PMMA NPs were produced via mini-emulsion polymerization. At first, 1 g magnetic modified NPs was added to 10 mL MMA under ultrasonics (80 W, 5 cycles with 2 min duration each) as the oil phase. SDS and CA, with a molar ratio of 1:3 (0.1 g SDS and 0.288 g CA), were dissolved in 100 mL DI water as a water phase. Then, the two phases were added together in the presence of ultrasonic treatment and N2 to form a mini-emulsion oil in water system, and the resulting mini-emulsion was placed in a four-necked-flask in a water bath, with the flask equipped with an N2 inlet, mechanical stirrer, and condenser. It was agitated for 30 min (400 r/min) and the temperature increased gradually to 60°C; 1 g benzoyl peroxide (BPO) was then added as an oil phase initiator to the flask and the polymerization proceeded under stirring for 5 h at 75°C. In the end, a brown product was separated by a strong magnet and washed with acetone 2 times and DI water 3 times under an external magnetic field until the remaining water became clear and the pH of the remaining media was 7.
Preparation of (Fe3O4/PMMA)-SiO2 nanoparticles
Before the silica coating of the above prepared NMPs, the surface chemical groups on the nanospheres needed to be hydrolyzed. A solution mixture containing 0.1 gr Fe3O4/PMMA NPs, 100 mL methanol and 2 mL ammonia were sonicated under mild power (40 W, 2 cycles each with 2 min duration). After that, the mixture was refluxed for 2 h in a flask and then washed with DI water. The NP surfaces were prepared for silica deposition according to the Stober method. In the first stage, 150 mL of a water/ethanol (1:4) solution containing 0.1 gr of the obtained nanoparticles was sonicated under weak power (20 W, 2 min). After transferring to a three-necked-flask equipped with a stirrer and a condenser, the temperature was set to 40°C, and after that 6 mL ammonia and 5 mL TEOS were added dropwise (fractionally). Then, the solution was agitated (400 r/min) for 12 h at 40°C. Finally, the obtained nanoparticles were separated and washed with DI water several times under a magnetic field (each time after washing, the samples were separated from the magnet and shaken several times) until the pH of the remaining water decreased to 7.
Results and discussion
The overall preparation of the PMMA (as core)/SiO2 (as shell) nanoparticles which have Fe3O4 nanoparticles attached with raspberry-like morphology had three stages, as shown in Scheme 1. The first stage was the preparation of superparamagnetic hydrophobic Fe3O4 nanoparticles by incorporating oleic acid in the co-precipitation method. Adding OA fractionally in the synthesis process of MNPs led to nearly mono-dispersed hydrophobic Fe3O4 nanoparticles. The second stage was the polymerization of PMMA on the surface of the OA modified superparamagnetic Fe3O4 for their protection. The polymer’s encapsulation around the OA-modified Fe3O4 surface was feasible because of the interaction between the oleic acid chains on the Fe3O4 surfaces and the MMA monomers. In the last stage, the nanoporous silica shell was formed by hydrolysis and TEOS condensation on the surfaces of the hydrolyzed Fe3O4/PMMA superparamagnetic nanoparticles. The TEOS layer could be deposited with the Sol-Gel method on the hydrolyzed surfaces by the interaction of the hydrogen atoms of the TEOS monomers on the surface.
29
In the ultrasonication process in the final stage, in the TEOS droplets containing Fe3O4/PMMA nanoparticles some of the Fe3O4 nanoparticles were released from the PMMA encapsulation to the TEOS droplets, (because of the power of ultrasonic waves); hence, these nanoparticles located on the outer shell of silica and the raspberry-like morphology was prepared. The silica shell with a high surface area has sufficient space for drug loading
11
but the size of nanoparticles should be controlled in the range of the optimum diameter (50–200 nm).
40
After every step, the nanoparticles were washed while on a magnet and the washing procedure is shown in the box in Scheme 1. The complete process of synthesize of the (oleic acid-Fe3O4/PMMA)-SiO2 magnetic nanoparticles, the PMMA and SiO2 structure, and the method of washing after each step are shown.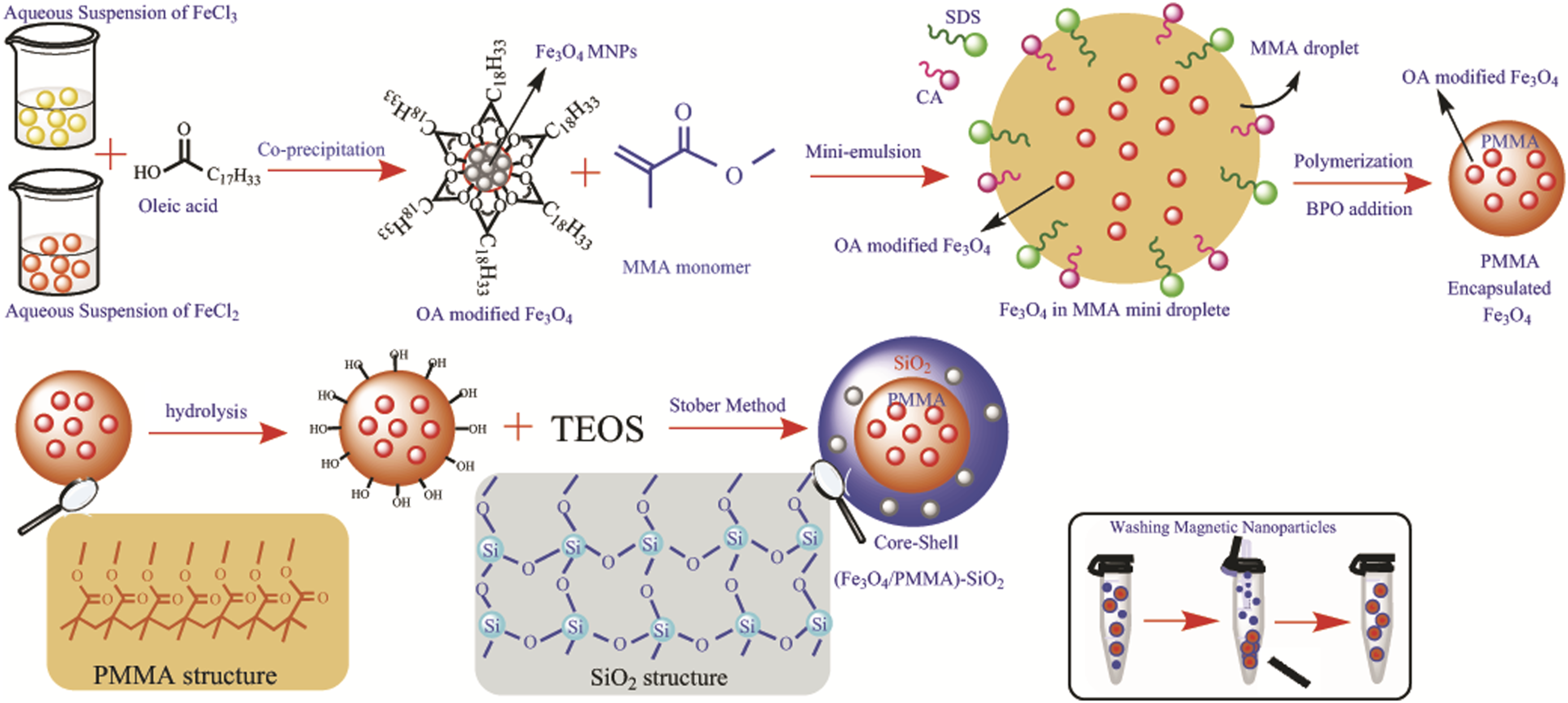
Morphological and structural characterization of the MNPs
In Figures 1(a) and (b) TEM images of the final nano-sized MNPs with narrow polydispersity are shown. Figure 1(a) represents the raspberry-like morphology distribution of the Fe3O4 nanoparticles (black dots) in both the PMMA (core) and SiO2 (shell), which shows the effective encapsulation of Fe3O4 nanoparticles by the polymer core and silica shell (black dots in gray surroundings). Figure 1(b) shows the spherical shape, narrow polydispersity and fine dispersion of the (Fe3O4/PMMA)-SiO2 nanoparticles. The polymer core encapsulates the hydrophobic OA-modified Fe3O4 nanoparticles, while the silica shell encapsulates the hydrophilic Fe3O4 nanoparticles which were released from the PMMA encapsulation due to using the ultrasonic waves, this is the reason for the formation of the raspberry-like morphology. Three regions of different electron densities confirm the formation of the dispersed Fe3O4 NMPs in the PMMA core and the outer shell of SiO2. The Fe3O4 nanoparticles, due to the electron beam absorption, appear as dark spots, while both the PMMA cores and Silica shells appear brighter. The average diameter of the nanospheres observed by TEM was approximately between 70 and 85 nm, and the core diameter of the PMMA/SiO2 was measured as <50 nm; thus, the thickness of the SiO2 layer was about 14 nm and the Fe3O4 nanoparticles were less than 10 nm in diameter. Also, SiO2 formed on the surface of the hydrolyzed Fe3O4/PMMA nanoparticles. The diameter of the PMMA core embedded hydrophobic Fe3O4 nanoparticles can become larger by increasing the PMMA to Fe3O4 weight ratio,
8
while the thickness of the SiO2 can be adjusted by the TEOS and ammonia content; an ultrathin silica shell can be obtained by using only a small amount of the aqueous droplets of TEOS and this methodology was used in this work. However, in order to have a high thickness coating, a large aqueous domain is essential.
41
In this article, an average thickness of a silica shell of 15 nm was observed. In order to avoid silica particles without the magnetic core, one of the effective strategies was adding the TEOS content dropwise,
41
and for preventing the formation of these core-free particles, hydrolysis of the nanoparticles before applying the Stober method was used. The Stober method is a sol-gel method that is used to prepare monodisperse silica microspheres; ammonia and tetraethyl orthosilicate (TEOS) are used as the catalyst and the silicon source, respectively.
42
Moreover, we did not find any silica nanospheres without the embedded Fe3O4/PMMA nanoparticles in our product (every PMMA (core)/SiO2 (shell) nanoparticle had magnetic NPs); this indicates the high yield of the OA-modified Fe3O4 nanospheres. Transmission electron microscopy images of the (oleic acid-Fe3O4/Polymethyl methacrylate (PMMA))-SiO2 magnetic nanoparticles.
An SEM image of the (Fe3O4/PMMA)-SiO2 nanoparticles is shown in Figure 2(a). The relatively broad particle size distribution and the spherical shape of these MNPs is shown in the image. It is noted, the SEM image has a larger size variation of the particles than the TEM image both much smaller (upper right) and larger (scattered) than 100 nm, which we attribute to the much larger number of particles imaged, with some of the imaged particles being aggregated. The polydispersity index (PDI) can be obtained from the DLS test through optical detection of the Brownian motion of nanoparticles in a liquid.
43
The PDI of (Fe3O4/PMMA)-SiO2 MNPs was about 0.101, which indicates the homogenous and uniform formation of MNPs in the aqueous medium. Besides this fact, the hydrodynamic diameters of the particles, as determined by the DLS test, was between 90 and 200 nm, and this narrow range represents the monodispersity of the MNPs. We note there was a difference between the hydrodynamic diameter of the particles from the DLS test and the diameter of particles measured in the TEM test. Also, the average hydrodynamic diameter of the OA modified-Fe3O4 was 10 nm (Figure 2(c)), and the particle diameter range was from 8 to 20 nm with a PDI of 0.14, indicating, the small size and monodispersity of the hydrophobic Fe3O4 nanoparticles. In the TEM test, because of how the sample was prepared and the nanometric scale of the test, we can observe the nanospheres separately depending on the magnification, hence, the observed size range was 60–90 nm with an average diameter of 79 nm. The SEM test is based on detecting the reflected or knocked-off electrons and it shows the surface morphology of the nanoparticles, but TEM creates images by electron transmission (passing electrons through the sample) and it shows the composition, morphology and size of the nanoparticles. We note the curves from the DLS tests represent the hydrodynamic diameter of the nanoparticles dispersed in water; their size was affected by the liquid medium and higher than the diameters calculated from the TEM test43,44; hence, the size of MNPs in these curves was from 90 to 200 nm. Based on these three supplementary tests the average diameter of single,
45
dry MNPs was 78 nm and the maximum hydrodynamic diameter, affected by the Brownian motions (random motion in a medium (liquid or gas) with particles suspended in it) in the water was about 200 nm. Scanning electron microscopy image of (OA-Fe3O4/PMMA)-SiO2 (a), Dynamic light scattering of (OA-Fe3O4/PMMA)-SiO2 composite nanospheres (b), and OA-Fe3O4 nanoparticles (c). OA: oleic acid.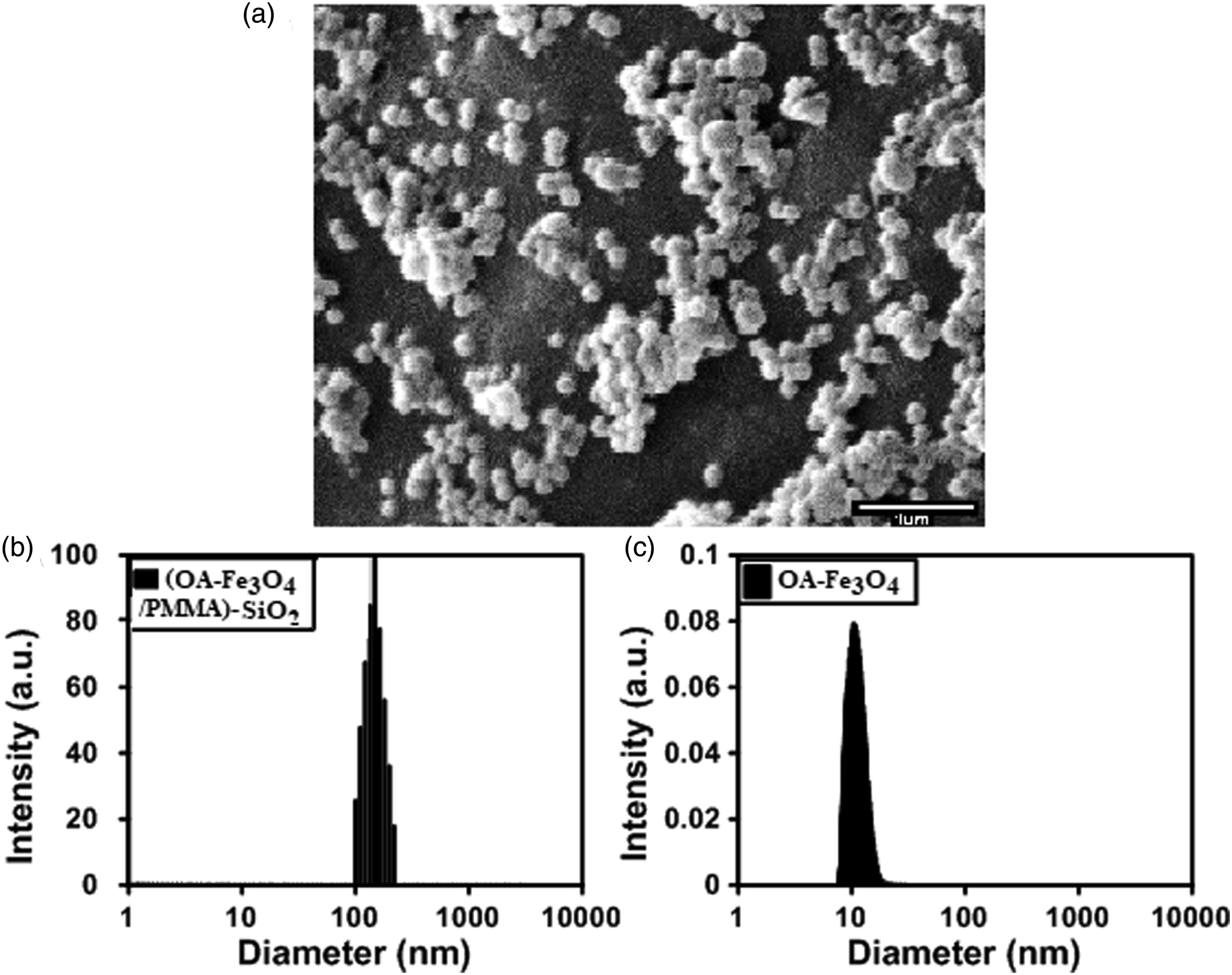
Microstructural study
FTIR spectroscopy was used to examine the as-synthesized Fe3O4-OA modified and Fe3O4/PMMA nanoparticles and the (Fe3O4/PMMA)-SiO2 composite nanospheres. Figure 3, curve a, shows the spectrum of the Fe3O4-OA modified nanoparticles; the absorption band at 573 cm-1 corresponds to the stretching vibrations of the Fe-O bonds and the double peaks near 2920 and the peak at 3433 cm-1 are related to the C-H groups of the OA segments. In the spectrum of Fe3O4/PMMA (Figure 3 curve b), the strong absorption band at 574 cm-1 is assigned to the Fe-O groups, which confirms the existence of Fe3O4. The new peak, at 625 cm-1, is due to the O-H and C-H groups of PMMA and the new peak at 1717 cm-1 is attributed to the ester bonds (-C(=O)OC-) of the PMMA, while the absorption bands at 2851 and 2920 cm-1 are due to the C-H groups of the PMMA as well as the C-H groups of OA and the absorption band at 3433 is assigned to the stretching of the O-H groups in PMMA. In Figure 3, curve c the peak at 574 cm-1 showed the Fe-O bonds and the peaks at 1717 and peaks at 2851 and 2920 cm-1 are assigned to the PMMA core. The broad absorption peak at 3433 cm-1 is due to O-H groups in the SiO2 coating due to condensation of incomplete silanol group (Si-OH) or absorbed water; four main characteristic SiO2 absorption bands were observed, Si-O-Si stretching, Si-OH stretching, Si-O bending and Si-O-Si bending
45
with their absorption peaks at 471, 801, 949, and 1095 cm-1, respectively. These peaks are due to the stretching vibrations of the SiO2 framework and terminal Si-O-Si (silanol) groups. Thus according to our FTIR analysis, magnetic Fe3O4 particles, PMMA and SiO2 existed in our nanoparticles. Fourier transform infrared spectra of (a) Fe3O4-OA modified nanoparticles, (b) OA-Fe3O4/PMMA nanoparticles, and (c) (OA-Fe3O4/PMMA)-SiO2 composite nanospheres. OA: oleic acid.
Thermogravimetric measurements
Figure 4(a) shows the TGA curves of the Fe3O4-OA modified/PMMA and OA modified-Fe3O4 NPs. The upper curve of Figure 4(a) shows the TGA of the OA-modified Fe3O4 NMPs; it is clear that the OA-modified Fe3O4 NMPs decomposed in 2 steps, the first mass loss, from 100 to 210°, might be due to the moisture evaporation (at 100°C) and free or loosely bound OA molecules (at a temperature between 170 and 210°C), and the second mass loss may be attributed to the OA chemically bound to the Fe3O4 NPs at temperatures between 280 and 42°C. The overall mass loss in this portion of the curve was about 12%. The lower curve shows the TGA curve of the Fe3O4-OA/PMMA nanospheres. The first mass loss from 100 to 250°C is again due to the moisture evaporation and loss of loosely bound OA molecules, as well as unreacted MMA monomers. The OA chemically bound with Fe3O4 NPs, based on the upper curve, decomposed between 320 and 420°C and the PMMA based on the excess decomposition, decomposed in the temperature range from 350 to 550°C, and the overall mass loss was about 19% at 550°C. The OA and PMMA mass loss peaks overlap because both of these materials have ester groups but according to the upper curve, the decomposition range of OA can be defined. In Table 1, the mass loss and the Fe3O4 content are listed. Based on the TGA curve, about 19% of the coating was the organic layer (OA+polymer), and 81% was the mineral content; here is the Fe3O4 content. In this research, this test was used as a validation for the VSM test because, through its results, the mineral content, which is here the Fe3O4 content, was obtained (Fe3O4 content is 81%), and by a simple equation (formula (1), defined in the magnetic characterization section below), the magnetic saturation can be calculated. a) Thermogravimetric analysis curves of OA modified Fe3O4 nanoparticles and OA-Fe3O4/PMMA nanoparticles and b) Vibrating sample magnetometry curves of Fe3O4 nanoparticles, OA-Fe3O4, OA-Fe3O4/PMMA nanoparticles, and (OA-Fe3O4/PMMA)-SiO2 composite nanospheres (b). OA: oleic acid. The magnetic measurement from TGA and VSM. TGA: thermogravimetric analysis; OA: oleic acid.

Magnetic characterization
Table of magnetic parameters of Fe3O4 NPs, (Oleic acid) OA-Fe3O4, OA-Fe3O4/PMMA, and (OA-Fe3O4/PMMA)-SiO2 nanospheres.
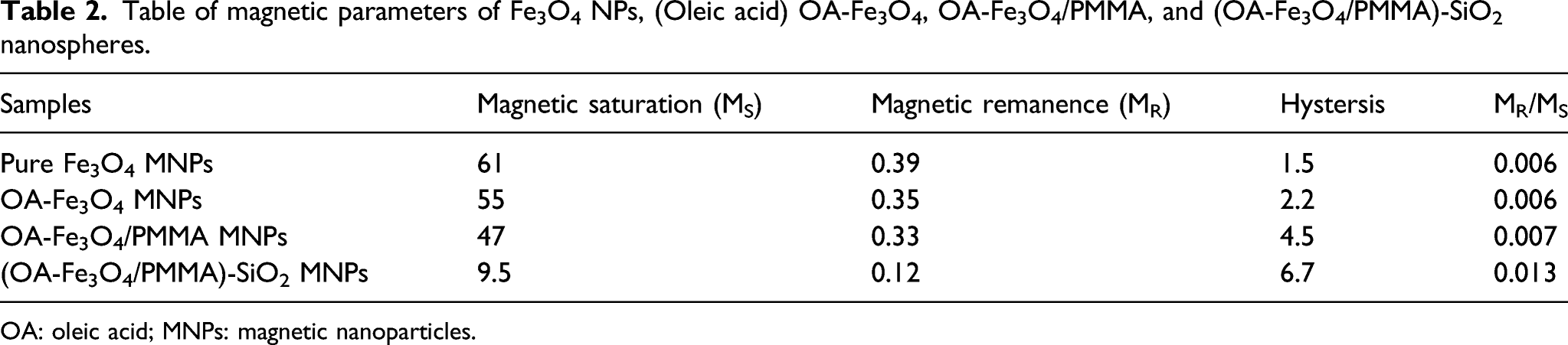
OA: oleic acid; MNPs: magnetic nanoparticles.
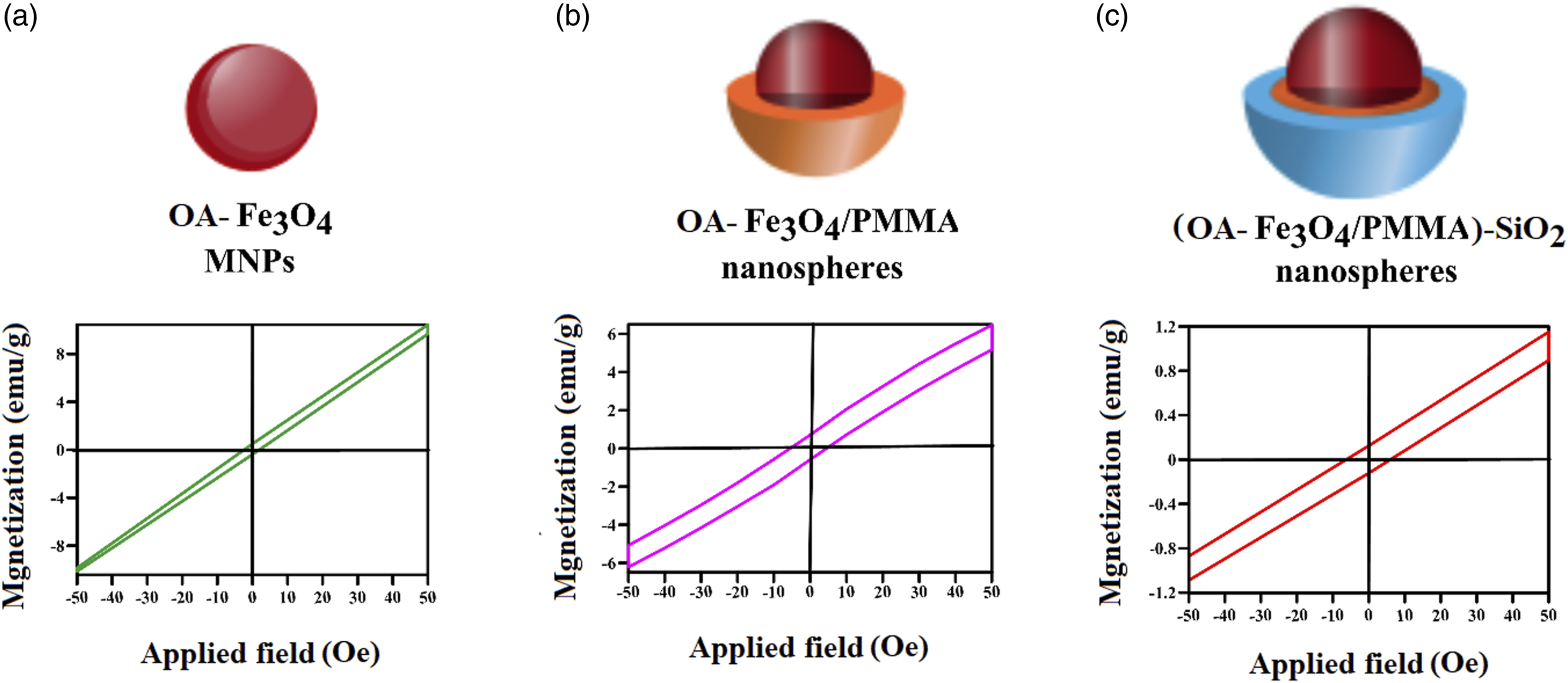
Magnified vibrating sample magnetometry plots of OA-Fe3O4, OA-Fe3O4/PMMA, and (OA-Fe3O4/PMMA)-SiO2 nanospheres, A, B, C, respectively. OA: oleic acid.
M. Das et al.
46
proposed that one could derive the Ms content from the mass loss percent in the TGA
Discussion
Our proposed synthesis process and details about the preparation of (OA-Fe3O4/PMMA)-SiO2 composite nanospheres are presented in Scheme 1. The Fe3O4 magnetic nanoparticles are hydrophilic, and they can disperse in the water medium. Poly (methylmethacrylate) is a hydrophobic monomer and, in mini-emulsion polymerization, it serves as an oil phase. In order to encapsulate the Fe3O4 with poly (methylmethacrylate), the Fe3O4 NPs were modified with oleic acid. Oleic acid can alter the surface characteristics of the Fe3O4 spheres and make them hydrophobic; it serves as a coating for the Fe3O4 NPs and an osmotic pressure agent for the formation of a stable mini-emulsion system.8,47 Modification with OA is the most critical stage in this procedure because it initiates all processes. Hence, for efficient modification, the nanoparticles should be modified homogeneously. We incorporated a facile way of modification in the conventional co-precipitation approach. If the modification process of the Fe3O4 NPs occurs accompanied by the nucleation and growth of the Fe3O4 nanospheres in the co-precipitation method, the Fe3O4 nanoparticles become hydrophobic and homogeneous in size. Another advantage of this kind of modification is that the nanoparticles are synthesized with a hydrophobic surface with a minimum size, about 6 nm, and with less aggregation of the Fe3O4 NPs. However, after the modification step, an excellent and facile way of washing with ethanol should be implemented to avoid an excess amount of OA; based on visual experiments, an extra amount of oleic acid causes inhomogeneity in the mini-emulsion, which is probably due to the formation of hydrophobic particles with inhomogeneous size. In other publications, in the synthesis of the Fe3O4 coated nanospheres, some Fe3O4 NPs without polymer coating existed, as shown by Yan et al. 22 ; also, the magnetic content of each composite particle differed. As shown in the TEM images in Figure 1, in our study free Fe3O4 nanoparticles not incorporated in the manufactured nanospheres were not observed, attributed to the significant modification of the Fe3O4 MNPs with OA and effective polymerization of the PMMA and SiO2 coverings on the Fe3O4 nanoparticles. Thus, our implementation of the modification with OA to avoid aggregation succeeded. In our mini-emulsion polymerization, because of the hydrophobic surface modification of the Fe3O4 NPs with oleic acid, the OA-Fe3O4 nanoparticles became dispersed in an oil phase; hence, ionic and nonionic surfactants could form the well-defined oil in water system containing the magnetic nanoparticles in the oil phase with a stable, narrow size distribution,48–50 this phenomenon is the basis for the formation of our monodisperse OA-Fe3O4/PMMA nanoparticles.
In our study, SDS (anionic surfactant) and CA (co-surfactant), 51 with a molar ratio of 1:3, were chosen as mixed surfactants for achieving system stability and narrow size distribution of the Fe3O4/PMMA nanospheres. The SDS chains located around the MMA droplets containing the OA-Fe3O4 nanoparticles and, because the CA co-surfactant is a fatty acid, it located in the oil phase (MMA droplets). Under ultrasonic force, the MMA monomer droplets containing the OA-Fe3O4 dispersed homogeneously in the water phase. Hybrid oil droplets with outer SDS and inner CA in the water phase formed by hydrophobic, solvophobic, and van der Waals forces. This stage is the most important and the sizes of the magnetic polymer composite nanospheres were determined in this stage. The initiator (BPO) was added after the gradual temperature increase to 60° and located in the oil phase; then, the Fe3O4 nanoparticles were covered with PMMA. In our research, we could synthesize separated, individual Fe3O4 nanoparticles encapsulated with PMMA and SiO2. The encapsulation by PMMA around the Fe3O4 was enough for stabilizing the Fe3O4 nanoparticles and preventing them from agglomerating.
For the next encapsulation by the Stober method with SiO2, a modification method needed to be implemented previously to alter the nanospheres' surface properties. The synthesized OA-Fe3O4/PMMA nanospheres have hydrophobic surfaces and the silica shell cannot deposit on such surfaces. Based on the report by Wang et al., 29 before silica deposition, we used a hydrolysis process to make the nanospheres hydrophilic; after the hydrolysis, the nanospheres can undergo interactions with TEOS by their -OH groups. Due to the PMMA coating of the Fe3O4 nanoparticles, there were many ester bonds on the surface of the nanospheres. These ester bonds reacted with hydroxyl groups due to catalysis of the base groups on the NH3OH and were hydrolyzed into the hydroxyl groups. 52 Hence, after hydrolysis, the carboxyl groups for silica deposition on OA-Fe3O4/PMMA nanospheres were present. After that, the best way for the synthesis of silica on the surface of nanoparticles is the Stober method because it produces a controllable and uniform covering; in this process, ethanol solvent was used to adjust the hydrolysis process and condensation with TEOS. A lower ratio of ethanol results in agglomerated silica particles; thus, for narrow size distribution, a sufficient proportion of ethanol should be employed.53,54 The resultant -OH groups act as alkaline groups, supplied by the ethanol solvent. 55 The silica shell is formed by hydrolysis and TEOS condensation, and this outer layer makes the magnetic nanoparticles stable to agglomeration. Silanol groups obtained by TEOS hydrolysis are absorbed on the hydrolyzed surfaces of the magnetic nanospheres by the reaction between the hydroxyl groups on the surface with silanol groups. The condensation process of the TEOS is needed for further silica deposition.29,55 Without the SiO2 layer, the MNPs tend to aggregate because of their dipole attractions. 55 Hence, a SiO2 shell with nanometric thickness is needed for stabilization and biomedical applications.
Conclusions
In summary, we successfully synthesized PMMA (core)/SiO2 (shell)-Fe3O4 nanoparticles with an average diameter between 70 and 90 nm. At first, we implemented a facile, modified co-precipitation method combined with oleic acid modification of the Fe3O4 to synthesize monodisperse, hydrophobic magnetic particles with a diameter of about 6 nm. After washing with ethanol to remove excess amounts of oleic acid, these magnetic particles were used in a mini-emulsion process for PMMA encapsulation to form the cores. After this step, the hydrophobic PMMA nanospheres’ outer surface was modified with ethanol and ammonia to permit the addition of a silica shell. The hydrophilic OA-Fe3O4/PMMA nanospheres were used in a modified Stober reaction to synthesize the SiO2 shell. In the PMMA encapsulation process, some of the Fe3O4 nanoparticles were released from the PMMA encapsulation due to the power of the ultrasonic waves, the silica shell encapsulates these released hydrophilic nanoparticles while the OA-modified nanoparticles stayed in the PMMA shell and this is the reason for the formation of the raspberry-like morphology. Given the excellent homogeneity, superparamagnetism, high magnetic content, and stability of the PMMA (core)/SiO2 (shell)-Fe3O4 nanoparticles, this new platform offers tremendous potential for magnetic particles with clinical application that can be used for magnetic resonance imaging contrast agents.
Footnotes
Acknowledgments
We wish to express our gratitude to Amirkabir University of Technology (AUT) and the Laboratory of Polymers and the Laboratory of Coating and Magnetic Molecules at Shahid Beheshti University for supporting this work.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.