
Abstract
The synthesis, characterization, and thermal properties of partially renewable poly(butylene terephthalate) copolyesters containing alditol units are described. These copolyesters were obtained by polycondensation in solution from mixtures of 1,4-butanediol and 2,3-di-O-benzyl-L-threitol with terephthaloyl chloride. Copolyesters with weight-average molecular weights oscillating between 4 000 and 12 000 g·mol−1 and dispersities around 1.5 were obtained. All them had a random microstructure and were thermally stable well above 300°C. Copolyesters containing up to 30% of dibenzyl threitol units were found to be crystalline and to adopt the same crystal structure as the parent homopolyester poly(butylene terephthalate). The melting temperature and crystallinity were observed to decrease, and the glass transition temperature to increase, with increasing amounts of alditol units incorporated in the copolyester. Furthermore, the crystallizability was depressed by copolymerization.
Introduction
Poly(butylene terephthalate) (PBT), is a semicrystalline polyester that is generally obtained by melt polycondensation of dimethyl terephthalate (DMT) with 1,4-butanediol (BD). PBT is of great interest for the manufacturing of vehicle components and other industrial applications such as electrical devices due to its good capacity to crystallize, its thermal and mechanical properties and its high resistance to the climatic changes. 1 Unfortunately, it is high resistant to degradation in aqueous media or by microorganisms which constitutes a great inconveniency for its use as material in provisional applications. Currently, great efforts are being made to make aromatic polyesters more hydrophilic and susceptible to hydrolytic degradation or biodegradation. 2 Copolymers containing aliphatic and aromatic counterparts are currently under study to elaborate decomposable materials able to satisfy new needs. 3
Recently, the incorporation of highly polar or hydrophilic moieties in the PET chain has proven successful in increasing its hydrolytic degradability. This approach includes the use of comonomers such as substituted terephthalic acids4–6 or polyhydroxylated diols7,8 derived from carbohydrates. The insertion of carbohydrate moieties in polycondensates such as polyamides and polycarbonates to obtain more hydrophilic and biodegradable polymers has been extensively explored in these last years.9–11 In the case of PBT, it has been copolymerized with different aldaric acids 12 and monocyclic or bicyclic acetalized sugars13–19 which were proved to produce polyesters with higher glass transition temperatures (Tg) enhanced hydrolytic degradability.
In this paper we report on the synthesis and characterization of a series of polyesters derived from PBT with variable contents of an alditol, 2,3-di-O-benzyl-L-threitol. This series permits estimating the influence of the amount of sugar moiety incorporated on the properties of these PBT copolyesters. Moreover, the deprotection of the benzyl groups in the polymer provides a suitable route to the preparation of PBT copolyesters bearing free hydroxyl groups. The structural characterization and thermal behavior of protected and unprotected PBT copolyesters were carried out. A comparative analysis of the detailed properties of those copolyesters is reported in this work.
Materials and methods
Common reagents and solvents were purchased from Sigma-Aldrich and used as received.
1H and 13C NMR spectra were recorded on a Bruker AMX-300 spectrometer at 25.0°C operating at 300.1 and 75.5 MHz, respectively. Polyesters and copolyesters were dissolved in a mixture of deuterated chloroform and trifluoroacetic acid (9:1), and spectra were internally referenced to tetramethylsilane (TMS). Here 10 and 50 mg of sample dissolved in 1 mL of deuterated solvent were used for 1H and 13C NMR, respectively. Sixty-four scans were acquired for 1H and 1000–10 000 for 13C with 32 and 64 K data points and relaxation delays of 1 and 2 s, respectively. Gel permeation chromatography (GPC) was carried out using 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) as the mobile phase at 35°C. GPC analysis was performed on a Waters GPC system equipped with a refractive index detector. Molecular weights were calculated against monodisperse poly(methyl methacrylate) standards using the Maxima 820 software. The thermal behavior of the polyesters was examined by differential scanning calorimetry (DSC) using a Perkin-Elmer DSC Pyris 1 calibrated with indium. DSC data were obtained from samples of 4–6 mg at heating/cooling rates of 10 or 20°C min−1 under nitrogen circulation. Thermogravimetric analysis (TGA) was carried out with a Perkin-Elmer TGA-6 thermobalance at a heating rate of 10°C min−1 under a nitrogen atmosphere. Powder X-ray diffraction patterns were recorded on flat photographic films with a modified Statton camera using nickel-filtered Cu Kα radiation with wavelength 0.1542 nm, and they were calibrated with molybdenum sulfide.
Synthesis of monomers
Diisopropyl 2,3-di-O-benzyl-L-tartrate
To a stirred suspension of sodium hydride (60% dispersion, 0.224 mol, 5.4 g) in dry THF (400 mL) was added dropwise a solution of diisopropyl-L-tartrate (20 g, 0.1123 mol) at 0°C. After being stirred for 1 h at room temperature, tetrabutylammonium iodide (TBAI) (8.5 g), a catalytic amount of 18-crown-6 (0.07 g) and benzyl bromide (26 mL) were successively added to the reaction mixture. The resulting suspension was stirred at room temperature for 24 h before being quenched by the addition of hydrochloric acid (1 N, 370 mL). The reaction mixture was diluted with water and extracted with ethyl acetate. The extract was washed with aq. NaHCO3 and brine, dried and concentrated to dryness. Recrystallization of the residue from methanol afforded to diisopropyl-dibenzyl-tartrate (75%) as colorless solid.
1H NMR (CDCl3, 300 MHz): δ (ppm) 7.30 (m, 10H, 2 Ph), 5.05 (m, 2H, 2 CH), 4.86–4.82 (d, 2H, CH2Ph), 4.49–4.45 (d, 2H, CH2Ph), 4.39 (bm, 2H, CH-CH3), 1.26–1.24 (d, 6H, CH3), 1.18–1.15 (d, 6H, CH3). 13C NMR (CDCl3, 75.5 MHz): δ (ppm) 170.91 (2 C=O), 137.65 (Ph), 128.72, 127.94, 127.52 (Ph), 80.53 (CH), 72.24 (OCH2Ph), 69.78 (CH-CH3) and 23.91 (CH3).
2,3-Di-O-benzyl-L-threitol
To a cooled solution of diisopropyl 2,3-di-O-benzyl-L-tartrate (10 g, 24 mmol) in dried THF (60 mL) under nitrogen atmosphere, LiAlH4 (97%) (2.4 g, 60.4 mmol) in dried THF (50 mL) was added and stirred for 12 hours. The mixture was cooled at 0°C, and H2O (6 mL), NaOH (15% w/v) (10 mL) and H2O (15 mL) were sequentially and slowly added. Then, the mixture was filtered and the solid extracted with hot acetone. The combined extracts were evaporated to dryness and the residue crystallized from methanol (white powder, 85% overall yield).
1H NMR (DMSO, 300 MHz): δ (ppm) 7.33 (m, 10H, 2 Ph), 4.64 (s, 4H, 2 CH2Ph), and 3.83–3.69 (m, 4H, 2 CH2, and m, 2H, 2CH). 13C NMR (DMSO, 75.5 MHz): δ (ppm) 144.57 (2C, 2 Ph), 133.26, 132.70, 132.37 (10 C, Ph), 85.70 (CH), 77.20 (2 OCH2Ph), and 66.65(CH2).
Synthesis of polymers PBxTByT: General procedure
The pure 2,3-di-O-benzyl-L-threitol or mixed with 1,4-butandiol (1.35 mmol) was charged in a round bottom flask provided with an inlet of argon. Dry 1,2-O-dichlorobenzene (8 mL) was added and the mixture was homogenized by stirring. Terephthaloyl chloride (274 mg, 1.35 mmol) was added under argon atmosphere. This solution was stirred for 24 hours at 140°C, 2 hrs at 170°C, 3 hrs at 180°C and 2 hrs at 180°C with vacuum. Finally, the residue was dissolved in a small volume of chloroform and the solution was added dropwise into cold diethyl ether (150 mL), where the polymer PBxTByT precipitated. The purification of the polymers or copolymers was carried out by redissolution in a small volume of chloroform (2 mL), and reprecipitation into diethyl ether. The pure polymer or copolymer (white powder) was dried under vacuum for 2 days and stored in a desiccator.
Hydrogenolysis of PBxTByT
In a stainless steel reactor provided with a 150 mL Teflon flask, PBxTByT (250 mg) was dissolved in methanol (50 mL), followed by the addition of palladium on carbon catalyst (Pd/C) (10%) in the amount of 10 mol-% of the benzyloxy groups. The reactor was purged with nitrogen, filled with hydrogen to a pressure of 50 Bar, and stirred at room temperature for 24 hours. Then, the catalyst was removed by filtration and the clean filtrate evaporated to give the partially deprotected polyesters (PBxTyT). The products were dried under vacuum at 30°C for 24 h.
Results and discussion
Synthesis and characterization
The preparation of the copolyesters was carried out by reaction of terephthaloyl chloride and a mixture of 1,4-butanediol and 2,3-di-O-benzyl-L-threitol in the selected proportions (Figure 1). For comparison purposes, the parent homopolyesters (PBT, PTBT) were synthesized by the same procedure from their respective pure diols. The polycondensation reaction proceeded in 1,2-dichlorobenzene as solvent (Figure 1). PBxTByT copolyesters were prepared from feed molar ratios of BD: alditol ranging from 90:10 to 50:50. The chemical constitution and composition of the resulting polycondensates were determined by 1H NMR (Figure 2), and their molecular weights were estimated by GPC (Figure 3). Data provided by these analyses are given in Table 1, where it can be observed that the copolyesters had compositions essentially similar to those of their respective feeds.

Synthetic route to PBxTByT and PBxTyT copolyesters.

1H NMR spectra of PTBT and PBxTByT copolyesters.

GPC chromatograms of PBT, PTBT and PBxTByT copolyesters.
Molecular weights, compositions, number-average sequence lengths, and randomness of the PBxBTyT copolyesters.

a Determined from the oxybutylene and oxythreitylene proton resonances observed in the 1H NMR spectra.
b Experimental values obtained from 13C NMR data and equations reported in Kint et al. 4 Theoretical values (in parentheses) were calculated on the basis of a Bernoullian dyad distribution using the copolyester composition data given in this table.
The microstructure of copolyesters was determined by 13C NMR analysis (Figure 4). These spectra showed single signals for the carbon atoms of tetramethylene units, whereas complex signals were observed for carbonyl and non-protonated aromatic carbons of the terephthalic units, indicating that these units are sensitive to sequence distribution effects. Thus, as shown in Figure 5, the resonance of the carbonyl and non-protonated aromatic carbons appeared as four signals respectively in the 167–167.9 ppm and 133.2–133.8 ppm chemical shift interval corresponding to the three types of dyads (BTTB or TBTB, BTB, and TBTTB) that are possible along the copolyester chain. The plot of the content in each type of dyad as a function of the copolyester composition reveals that the microstructure of the copolyester is statistical, with randomness quite near unity in all cases. The weight-average molecular weights of copolyesters were found to be roughly within the 4 000–12 000 g·mol−1 range.

13C NMR spectra of PTBT and PBxTByT copolyesters.
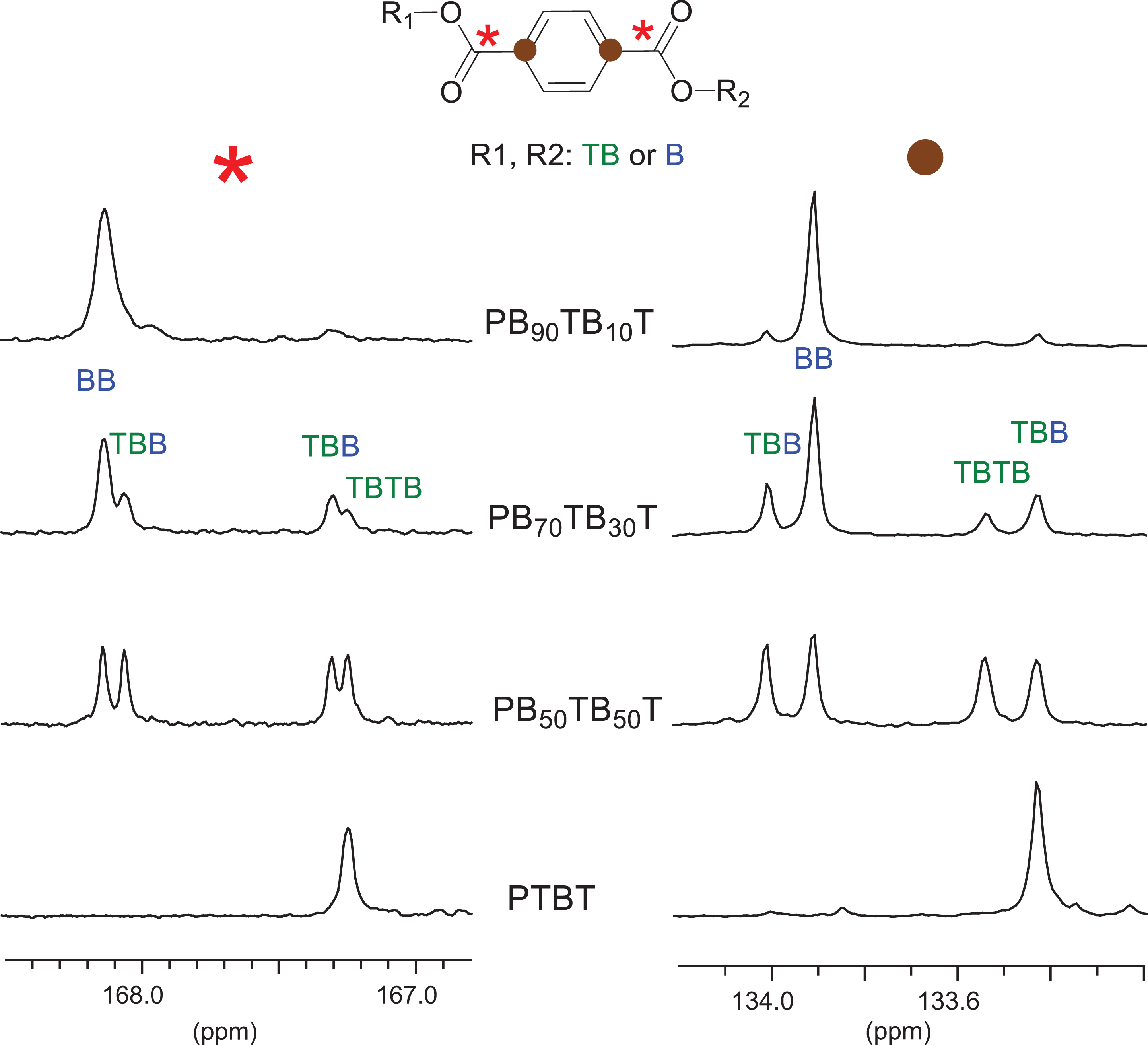
13C NMR spectra of PTBT and PBxTByT copolyesters in the carbonyl and non-protonated aromatic carbon region.
Thermal properties
The thermal behavior of copolyesters has been comparatively studied by DSC and TGA and the thermal parameters resulting from these analyses are given in Table 2. Thermogravimetric analysis reveals that all copolyesters are very stable with a thermal decomposition temperature above 400°C, slightly higher than for the PBT (Figure 6).
Thermal properties and X-ray spacings of polyesters.
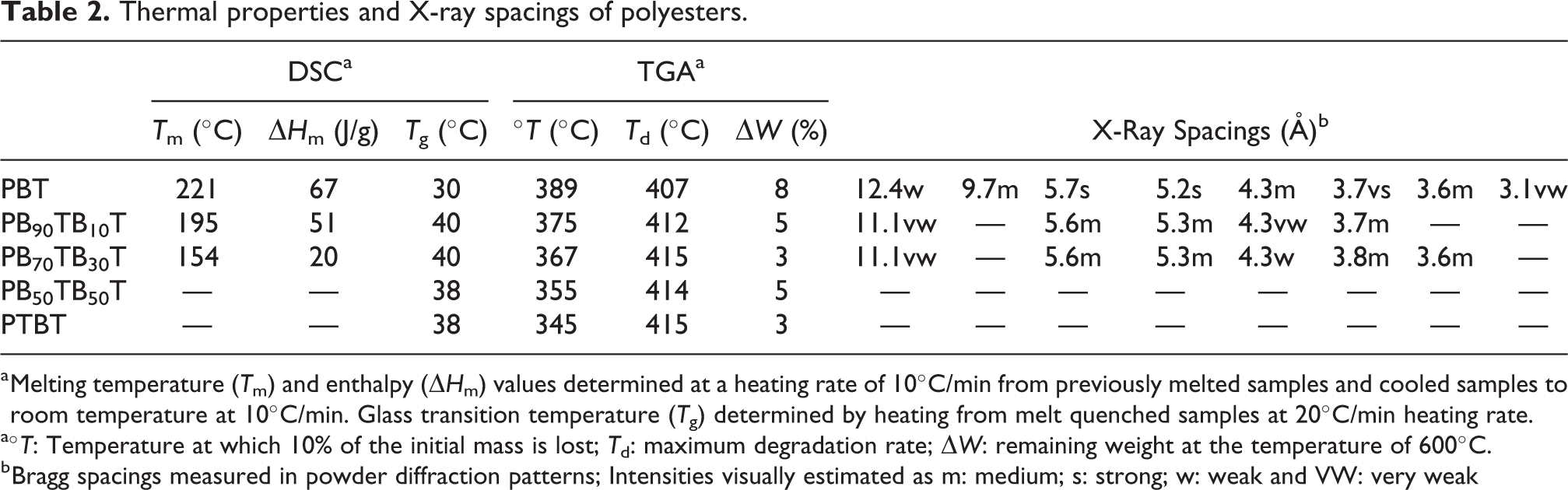
a Melting temperature (Tm) and enthalpy (▵Hm) values determined at a heating rate of 10°C/min from previously melted samples and cooled samples to room temperature at 10°C/min. Glass transition temperature (Tg) determined by heating from melt quenched samples at 20°C/min heating rate.
aºT: Temperature at which 10% of the initial mass is lost; Td: maximum degradation rate; ▵W: remaining weight at the temperature of 600°C.
b Bragg spacings measured in powder diffraction patterns; Intensities visually estimated as m: medium; s: strong; w: weak and VW: very weak

TGA thermograms of (a) PTBT and PBxTByT copolyesters (b) PB70TB and its DTG curve.
The DSC analysis reveals that the insertion of the 2,3-di-O-benzyl-L-threitol in the main chain of PBT results in a noticeable decrease on melting temperature and enthalpy and an increase in the glass transition temperature from 30°C to 40°C. As shown in Figure 7, well-defined melting peaks, indicative of the presence of a crystalline phase, were observed for copolyesters containing up to 30% of threitol units. X-ray diffractions results were in full agreement with DSC observations; the most characteristic Bragg Spacings measured for each polymer are compared in Table 2. X-ray data revealed that the triclinic crystal structure of PBT is retained in the copolyesters (Figure 8).

DSC thermograms of PBT, PTBT and PBxTByT copolyesters recored from crystallized samples from the melt (left) and melt quenched samples (right).

Powder WAXD profiles of PBT and crystalline PBxTByT copolyesters with indication of
Hydrogenolysis of copolyesters
Hydrogenation of the benzyl ethers, which is a common reaction extensively used for removing the benzyl group, required severe reaction conditions to proceed in this case (Figure 1). Rather high amounts of catalyst and the application of fairly high pressures were needed to obtain significant conversion. The preparation of the hydroxylated copolyesters was carried out following the same procedure used previously by us. 20
The resulting copolyesters were obtained in about 80% yield and they have weight-average molecular weights between 4000 g·mol−1 and 12000 g·mol−1 (Table 3). No variations of the molecular weights were observed for these hydrophilic copolyesters which indicates that no breaking of the main chain took place along the deprotection reaction.
Molecular weight, composition and thermal properties of hydrogenated copolyesters.

a Determined from the benzyl and oxybutylene proton resonances observed in the 1H NMR spectra.
b Melting temperature (Tm) and enthalpy (▵Hm) values determined at a heating rate of 10°C/min from previously melted samples and cooled samples to room temperature at 10°C/min. Glass transition temperature (Tg) determined by heating from melt quenched samples at 20°C/min heating.
cºT: Temperature at which 10% of the initial mass is lost; Td: maximum degradation rate; ▵W: remaining weight at the temperature of 600°C.
The 1H-NMR spectra of PB70TB30T and its hydrogenated form are shown in Figure 9 and reveals, in all cases, a partial debenzylation of the copolyesters studied. The signals characteristics of the benzyl side groups decrease significantly indicating that these groups were partially converted to hydroxyl groups. These signals were used for quantification of the degree of hydroxylation. Signals due to hydroxyl groups could not be quantified due to its rapid interchange with the acid protons of the solvent used for acquiring the NMR spectra. The results indicated that the degree of free hydroxyl group was found to increase with the content of the 2,3-di-O-benzyl-L-threitol units in the PBT chain (Table 3).

1H NMR PB70TB30T and partially debenzylated PB70T30T copolyester with peak assignments.
Thermal transition parameters of partially hydroxylated copolyesters were measured by DSC and they are collected in Table 3. Surprisingly the three copolyesters appeared to be crystalline generally with an increase in all parameters with the content of both protected and unprotected side groups (Figure 10(a)). In all cases, the degree of conversion from protected to unprotected side chain was found to be about 70%, which means that the changes taking place in thermal properties cannot be related to the degree of the conversion but probably to the microstructure of these copolyesters resulting from the hydrogenation.

(a) DSC thermograms of PBxTyT copolyesters. (b) TGA thermogram of PB70T30T.
The thermal stability of the copolyesters was slightly affected by the hydrogenation reaction. An illustrative TGA profile of PB70T30T is shown in Figure10(b). The onset decomposition appeared above 300°C indicating that the partially unprotected side chain copolyesters have similar stability to those having the hydroxyl group protected. In all cases, a single decomposition peak was observed at around 410°C.
Conclusions
Preparation of PBT copolyesters containing up to 50% of threitol units derived from naturally occurring tartaric acid is possible by polycondensation in 1,2-dichlorobenzene solution.
The copolyesters have relatively high molecular weights and a random microstructure. They are semicrystalline up to contents of 30% threitol units.
The melting temperature of PBT is considerably depressed by the presence of the threitol units, but its thermal stability is scarcely affected, being stable up to 400°C in all cases.
The debenzylation of three copolyesters was carried out by hydrogenation and lead to the corresponding partially hydroxylated copolyesters with a degree of conversion of about 70%. They appeared to be semicrystalline with an increase in all parameters as the degree of debenzylation increases. The thermal stability of the copolyesters was slightly affected by the hydrogenation reaction.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.