
Abstract
Hybrid films of poly(3-hydroxybutyrate) (PHB) and modified cellulose by solution casting method were prepared, aiming to study the influence of modified cellulose (0.25%, 0.5% and 0.75% w/w) on the structural organization and thermal properties of PHB matrix. The modified cellulose showed good dispersion in polymer matrix, due to the high compatibility between phases explained by similarity of polymer and filler structures. The composites were studied by wide-angle X-ray diffractometry, differential scanning calorimetry, thermogravimetric analysis, and time-domain nuclear magnetic resonance (NMR) spectroscopy. The conjugation of results coming from those techniques allowed to determine the cellulose concentration that had the highest influence on crystalline planes and degree of crystallinity of PHB, that is, the influence in the composite structure. The molecular dynamics obtained by NMR showed a reciprocal influence between polymer matrix and cellulose particles, enhancing the interactions present in the agglomerated particles.
Introduction
The extensive use of petrochemical-based polymeric materials has led to significant environmental impact, mainly regarding waste disposal. Consequently, the search for biodegradable materials is among the biggest challenges for scientific community during the 21st century. Tissue engineering, drug carriers, and food packaging are examples of applications that require constant study and new developments in this area. 1,2
The polyhydroxyalkanoates (PHAs) are linear-chain polyesters, yielded by microorganisms as a mean to store carbon and energy. They are biodegradable, which makes them interesting materials from the point of view of sustainability. 2,3 Besides this important feature, due to similarities of mechanical properties, PHAs are often used to replace highly stable synthetic polymers, which take long time for degradation after disposal. 4 The poly(3-hydroxybutyrate) (PHB) is a thermoplastic polymer from the PHAs family, having crystalline melting temperature (T m) around 175°C, glass transition temperature (T g) of 5°C, and tensile strength similar to polypropylene. 5,6 These properties are directly related to the morphology and crystallinity degree of the PHB, which define the mechanical properties and natural degradation rates. 7 Differently from other PHA polymers, it has lower thermal stability and is more brittle. The thermal instability of PHB at temperatures above its melting point restrains the artifacts fabrication that involves melting, like packages. 4,8 -12 The disadvantages of PHBs can be diminished by the addition of organic or inorganic particles that can bring different properties to the polymer when well dispersed and distributed in the matrix. Arrieta et al. 13 managed to improve the processability of the PHB through the incorporation of PLA. 7,8,12 -15
Cellulose is a natural polymer present in plants, being the most abundant polymer in the planet. Its use is preferred if compared to polymers of fossil precedence due to multiple reasons, including biodegradability, stiffness, thermal stability, abundance, ease of exploitation, and biocompatibility 16 ; these characteristics can enhance the properties of PHB polymer matrices. The use of cellulose brings improvements to mechanical and thermal properties when compared to pristine polymer matrix. 14,16,17 It is a known fact that cellulose nanocrystals (CNCs), when added in low proportions in polymer matrix, are able to provide the aforementioned improvements, since they have higher contact surface area than the microcrystalline cellulose. 2,11,12,18,19 Nonetheless, Dasan et al. 19 reported an agglomeration tendency of the CNCs at high concentrations, which occurred due to strong intermolecular interactions of OH groups in these structures; and accordingly, either the modification of the OH groups or the use of compatibilizing agents should be considered. Ten et al. 18 evaluated the compatibilization effect of polyethylene glycol in the PHB/CNC nanocomposites; they observed improvements in mechanical properties with 5% of CNC w/w concentration. The presence of hydroxyl groups allows many modifications in cellulose, decreasing the particles’ hydrophobicity and improving its interfacial interaction with the polymer matrix. 13,17 Lin et al. 20 and Goffin et al. 21 worked on cellulose modification, studying its influence in PLA and PCL structure.
Pure PHB and cellulose-PHB hybrid films were prepared by solution casting method, aiming to obtain greener materials for current sustainability scenario. The behavior of the obtained materials was analyzed by wide-angle X-ray diffractometry, thermogravimetry, differential scanning calorimetry (DSC), and time-domain nuclear magnetic resonance (TD-NMR).
Experimental section
The polymers used in this study were supplied by Biocycle (PHB Industrial S/A, Serrana, São Paulo, Brazil), while the microcrystalline cellulose was purchased from Synth (Diadema, São Paulo, Brazil). Glacial acetic acid was purchased from Quimex (Uberaba, Minas Gerais, Brazil). Sulfuric acid and chloroform were supplied by Sigma-Aldrich (São Paulo, Brazil). All materials were used without further purification.
Modification of cellulose
The modification and characterization of microcrystalline cellulose was made according to the procedure of Melo et al. 22 in a previous work, in which they performed the surface modification using a 3% w/w solution of microcrystalline cellulose in concentrated acetic acid with sulfuric acid as catalyst. The reaction was carried out with the assistance of a mechanical stirrer (IKA RW 20 digital, Campinas, São Paulo, Brazil) and ultrasound (Eco-sonics Q3.0/40A, 40 KHz frequency, Indaiatuba, São Paulo, Brazil) with heating, during 20 h of acid hydrolysis. The obtained solution was centrifuged at 12,000 × g and washed 10 times with deionized water. Afterward, the material was kept for 48 h in the oven to achieve complete solvent elimination. A detailed discussion about microcrystalline cellulose modification with acetic acid can be found in the literature. 22
Preparation of PHB/cellulose films
The production of PHB (Biocycle) (Mn 147.596 g mol−1, Mw 376 g mol−1, and PDI 2.5) films with cellulosic filler was made in a three-step solution. The previously modified cellulose was dispersed in chloroform (EMSURE® ACS, Brazil) using an ultrasound equipment (Ultronique QR500, 20 KHz frequency, Eco-sonics, Indaiatuba, São Paulo, Brazil) for 3 min and maximum power, at concentrations of 0.25%, 0.50%, and 0.75% (w/w). Then, it was added the PHB in proper amount to obtain solutions at 5% w/w. The system was kept under magnetic stirring for 24 h. Afterward, the solutions were heated at 50°C and kept under magnetic stirring for 1 h to complete the PHB solubilization. The solutions were cooled to 25°C and put once more through ultrasonic dispersion cycle for 1 min. After the dispersion process, the mixture was poured into Petri dishes to evaporate the solvent. All characterization was performed on dry films.
The wide-angle X-ray diffraction (WAXD) analysis was performed in films using Rigaku Miniflex diffractometer (Japan) with CuKα radiation (λ = 1.5418 Å) at experimental conditions of 40 kV, 20 mA, step of 0.05° s−1, considering 2θ from 2° to 60° and resolution of 2°. The crystallinity index of the materials was calculated through the Ruland–Vonk method 23 using the Fityk 1.3.1 software.
Thermogravimetric analysis (TGA) was performed using TA equipment model Q500 (USA). The temperature range was from 40°C to 350°C, at heating rate of 10°C·min−1, under nitrogen atmosphere. Mass loss 10% (T 10%), maximum (T max) and endset (T endset) degradation temperatures were measured.
DSC analyses were performed using a TA equipment Q1000 model. The films were analyzed under nitrogen atmosphere, according to the following cycles: in the first cycle, the sample was heated from −25°C to 200°C at a heating rate of 10°C·min−1, keeping the material at 200°C for 1 min; the second cycle was a cooling at high rate—quenching—until −25°C; at the third cycle, the same temperature range and heating rate of the first cycle were applied; the fourth cycle was performed at a cooling rate of 10°C·min−1 until −25°C; and at the fifth cycle, the same temperature range and heating rate of the first cycle were applied. T g, the crystallization temperatures in the cooling (T cc) and the heating (T hc), T m, and the crystallization (ΔH c) and melting (ΔH m) enthalpies were assessed. The degree of crystallinity was calculated according to equation (1)

where ΔH
m is the measured melting enthalpy and
TD-NMR technique studies the molecular dynamics related to the regions of higher stiffness—restriction in the mobility of hydrogen molecules—which shows higher relaxation times, while the more flexible regions are able to relax in shorter times. The measurements were performed in a Maran Ultra spectrometer with an electromagnetic field of 0.54 T (operating at a frequency of 23.4 MHz for the proton), with internal tube diameter of 18 mm, at 30°C. The excitation pulse 90° was automatically calibrated to 7.5 µs of duration. The longitudinal (T 1) relaxation times were measured employing the inversion–recovery pulse sequence. The experiment was conducted exploring a list of recovery intervals of 40 values logarithmically spaced from 0.1 ms to 20,000 ms, with recycle interval of 5 s and four accumulations.
Results and discussion
Structural characterization
The X-ray diffractograms of pristine PHB and composites films are shown in Figure 1. The PHB has an orthorhombic unit cell, characterized by crystalline planes (020), (110), (021), (101), (111), (121), (040), and (222), ascribed to the 2θ reflection angles = 13.5°, 16.8°, 19.8°, 21.4°, 25.6°, 27.2°, 30.2°, and 44°. 2,24

XRD diffractograms of pristine PHB and composites. XRD: X-ray diffractometry; PHB: poly(3-hydroxybutyrate).
Peaks of composites (0.25% and 0.5% cellulose) presented a slight shift to higher angles, in agreement with report by Yu et al., 25 indicating that there was a decrease in the distances between the crystalline planes in the presence of cellulose, probably due to the formation of smaller crystals. On the other hand, the peaks of the 0.75% cellulose composite were centered in the same position as PHB lattice, probably due to the new intermolecular interactions and organizations. The sample with 0.50% cellulose showed an increase in peaks intensity, as the filler content was increased, which possibly led to modification of crystalline packing of polymer chains, enhancing crystal growth. 2 The 0.75% cellulose composite presented intensity diminution of diffraction peaks when compared to 0.50% composite, which could be explained in the reaching of a dispersion limit, over which the presence of cellulose hinders the packing of PHB long chains; as a result, the agglomeration could lead to entanglements or physical nodes, damaging the dispersion of the filler in the matrix.
In the crystalline planes (020) and (110)—corresponding to the α crystals of the helical conformation chain—it can be noticed a narrowing of the peak bases, probably caused by higher interlamellar distance homogeneity in these planes. This narrowing indicates a better structural organization of PHB crystals due to the addition of cellulose in the studied concentrations, which is in agreement with results reported by El Hadi. 5 Planar zigzag conformation plane at 2θ = 20° (Figure 1) did not present any significant change. 1,10
Table 1 shows the crystallinity degree indices of PHB and its composites. Cellulose incorporation caused an increase in the crystallinity degree, bringing changes in the chains packing and crystalline morphology of the PHB matrix. Despite the increase in the intensity of some peaks of neat PHB, the 0.25% composite did not change the degree of crystallinity, while the samples with 0.50% and 0.75% showed the highest values. For the 0.75% sample, it is observed a reduction in the value if compared to the sample with 0.50%, probably because in this concentration, the cellulose hindered the chains packing influencing thus the morphology of the crystals, as already observed in the diffractograms shown in Figure 1.
Degree of crystallinity (X c) of PHB and composites.

PHB: poly(3-hydroxybutyrate).
Thermal properties of composite
The TGA/derivative thermogravimetry (DTG) curves are shown in Figure 2(a) and (b), respectively. The neat PHB degraded in a single step, as seen by DTG curves. According to the literature, the weight loss at 160–220°C is attributed to random chain scission has occurred by cis-elimination, giving byproducts like crotonic acid and related oligomers. 7,26 The presence of a shoulder at the derivative peak was noticed for the 0.75% composite, apparently due to the large amount of cellulose, as observed by Patrício et al. 6

(a) Degradation curve and (b) derivative curve of PHB and its cellulose-formed hybrids. PHB: poly(3-hydroxybutyrate).
There was an increase in the T max value (271°C and 280°C) of the systems containing 0.25% and 0.50% when compared to neat PHB (267°C), and then a decrease (262°C) for the 0.75% system. The same tendency was observed in T 10% and T final values (Table 2). The increase in thermal stability along with the addition of esterified cellulose to the PHB matrix is explained by the compatibility between carbonyl and acetate groups in the modified cellulose surface. The same type of interaction was described by Yuwawech et al. 27 Thus, the cellulose effect in the polymeric matrix depends on the reinforcement and concentration, which will be reflected in the dispersion and distribution of the cellulose, due to the strong hydrogen interactions between the PHB carbonyls and cellulose hydroxyls. 25
Degradation temperatures of composites at 10% mass loss (T 10%), maximum degradation rate (T max), and endset (T endset).
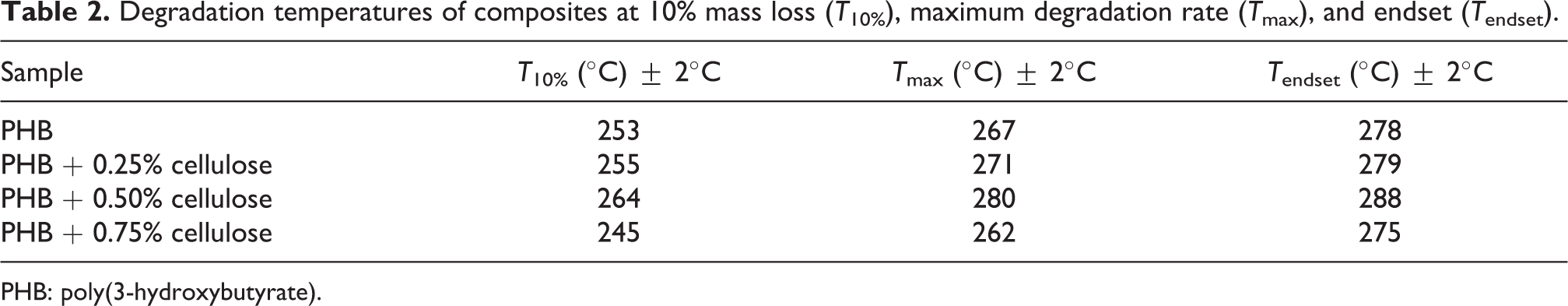
PHB: poly(3-hydroxybutyrate).
The insertion of acetate groups on cellulose surface did not alter the degradation temperature of PHB matrix even at higher concentrations, which differs from work reported by Yu et al., 25 who investigated the influence of cellulose prepared by different hydrolysis methods in Poly(3-hydroxybutyrate-co-3-hidroxyvalerate) (PHBV) matrix and verified that sulfate groups detach easily because of its lower energy, which decreases the composite thermal stability.
Figure 3 shows the third heating cycle DSC curves for neat PHB and PHB/cellulose composites. PHB has two characteristic endothermic peaks, related to crystalline melting. The crystals that melt at a lower temperature are smaller and/or more imperfect, formed in the drying process of the material before the heating. 28

DSC curves (third heating) of the PHB and PHB/cellulose composites. PHB: poly(3-hydroxybutyrate); DSC: differential scanning calorimetry.
A crystallization event was noticed during the heating cycle for the composites with 0.50% and 0.75% cellulose, which was neither observed for neat PHB nor for the composite with 0.25%. The presence of cellulose also caused changes in the population of crystals that melt at a lower temperature; there was a decrease in the intensity of the melting event related to those crystals, indicating that the presence of a high concentration of cellulose did not affect the crystals growth, favoring the formation of more perfect PHB crystals, which is in accordance to literature report. 12,13,28
The incorporation of 0.25% and 0.50% cellulose had no significant effect in the X c when compared to neat PHB. At the 0.75% concentration, there was a decrease of X c, probably because at this concentration the cellulose was interfering with the PHB crystallization process, as observed in the WAXD analysis and in the Table 3. The data from the first and third heating scans were similar, which corroborates the discussion and comparison with WAXD data.
Glass transition temperature (T g), melting temperature (T m), cooling (T cc) and heating crystallization temperature (T hc), and degree of crystallinity (X c) of the materials.
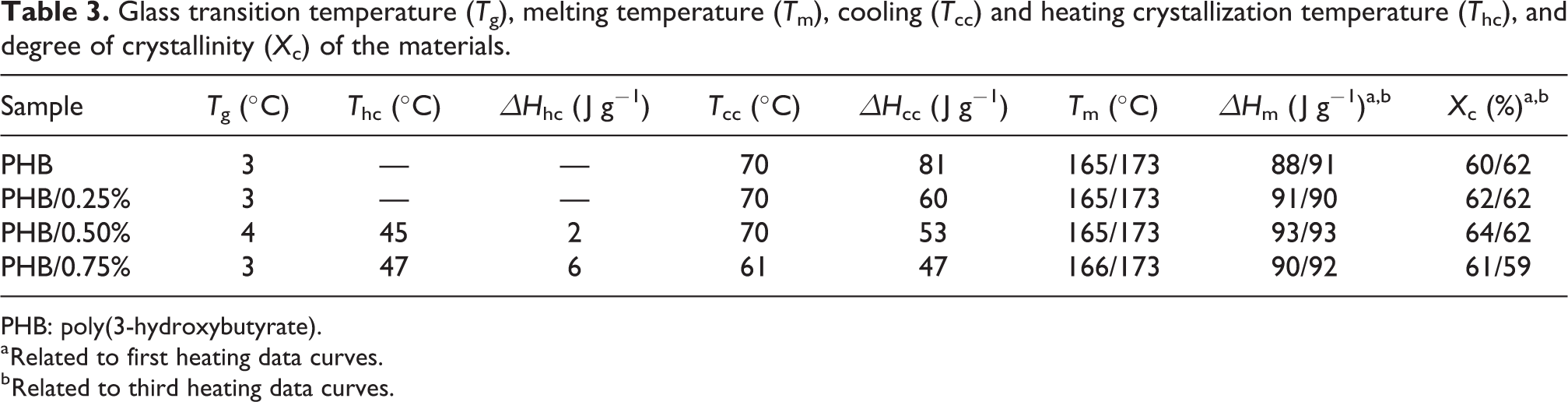
PHB: poly(3-hydroxybutyrate).
a Related to first heating data curves.
b Related to third heating data curves.
In Figure 4, it can be observed that the addition of cellulose has broadened the cold crystallization peak bases, indicating that there was higher heterogeneity in the formed crystals. Other studies have already shown the effect of heterogeneous nucleation provoked by the addition of cellulose in PHB and PHBV matrix. 5,12,29 In composites containing up to 0.50% cellulose, there was no shift of the central point of the peak, although it was observed to lower temperatures for the sample containing 0.75%, occurred due to the high amount of cellulose present in the matrix, which disrupts the organization of the chains and makes them require more time for chain alignment and consequent crystallization.

DSC second cooling curves of the obtained PHB/cellulose composites in different concentrations of modified cellulose. PHB: poly(3-hydroxybutyrate); DSC: differential scanning calorimetry.
Molecular dynamic analysis through TD-NMR
Table 4 shows the values of the spin-lattice relaxation times (with time constant, T1 H) of materials. The relaxation times determined by one exponential provide the mean T1 H values, related to the overall behavior of the samples, which are influenced by chemical structure, molecular interactions, and polymer organization, depicting the contribution of all relaxation times. 30 It has been observed that the addition of 0.25% cellulose caused a reduction in the value of T1 H when compared to pristine polymer. There was an increase in the molecular mobility due to the possible insertion of cellulose among the polymer chains, causing their separation. This behavior was also noticed by Santos and Tavares 31 when using cellulose concentrations higher than those used in the present work. This increase in the molecular mobility also indicates weak interactions between the polymer chains and cellulose, since the separation between chains allowed them to move with more freedom. 32
Relaxation times of hydrogen nuclei of PHB and PHB/cellulose composites.

PHB: poly(3-hydroxybutyrate).
The material containing 0.50% cellulose showed higher T1 H value, showing that this cellulose content caused greater rigidity. This was probably due to the formation of strong intermolecular interactions that restricted the molecular mobility of hydrogen groups. The same has occurred to the material containing 0.75%, which also showed a slight decrease in the relaxation time compared to neat PHB.
Through deep analysis of relaxation times with two exponentials, two relaxation values (in regions of greater and lower mobility) and their respective percentage contributions can be identified. The PHB has a T1,1 H of 7 ms, referring to the absorbed humidity, which has freedom of hydrogen molecules movement, and a T1,2 H of 612 ms, ascribed to the regions where hydrogen movement is more restricted. The incorporation of 0.25% cellulose into the PHB matrix caused a slight increase in the value of T1,1 H and a large decrease in the value of T1,2 H, which can be related to higher freedom of polymer chains movement.
For the 0.50% cellulose sample, it is observed an increase in both T1,1 H and T1,2 H values. This result indicates a change in the organization of the material due to good dispersion and formation of strong intermolecular cellulose–polymer interactions, which leads to the limitation of the hydrogen movements and made the sample more rigid. When compared to the neat polymer, the material with 0.75% cellulose showed an increase in the two relaxation times; when compared to the sample with 0.50%, there was a decrease in relaxation times; this can be ascribed to an insufficient dispersion of the cellulose at this concentration (0.75%), which might have caused the agglomeration of the filler, 33 and the agglomeration has increased the free volume around cellulose particles, granting more freedom to hydrogen linked to the closer polymer chains and reducing relaxation time.
Figure 5 shows the domain distribution curves of pristine PHB and composites relaxation. It was observed the narrowing of the lower mobility domain peak bases as the cellulose concentration increased to 0.50%; those samples showed more homogeneous behavior of hydrogen nuclei relaxation times, due to the good dispersion and interaction with the matrix. There was a large widening in the peak base of the 0.75% sample, implying a greater heterogeneity in the relaxation behavior of hydrogen nuclei. Apparently, the dispersion of cellulose at this content furthered different forms of organization and interactions, which may have led to greater heterogeneity.

Domain distribution curves of relaxation times for neat PHB and composites. PHB: poly(3-hydroxybutyrate).
Conclusion
Hybrid films of PHB and modified cellulose were prepared by solution casting method. Among the studied concentrations, the 0.25% and 0.5% w/w were those which showed better interaction with the polymer matrix, enhanced by the alteration of the crystalline planes and degree of crystallinity seen through WAXD analysis. The TGA showed an increase in T 10% temperature up to 0.5% of cellulose, and then a decrease of this parameter in 0.75% cellulose, indicating the poorest filler dispersion in the matrix. The NMR analyses corroborated these results. The relaxation times with two exponentials showed an increase in relaxation times of the greater mobility and lower mobility region, indicating a change in material organization and the formation of strong intermolecular cellulose–polymer interactions at 0.25% and 0.5% w/w cellulose. The DSC results showed that cellulose had no nucleating effect on PHB; however, it has influenced the chains packing, making the crystals more heterogeneous.
Footnotes
Acknowledgement
The authors would like to thank the Brazilian agencies CNPQ and FAPERJ.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was financed in part by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior, Brasil (CAPES), Finance Code 001.