
Abstract
This study aims to investigate the potential of two local fibers, namely El Diss and El Retma, which are abundant in the mountains of North Africa (Sétif, Algeria), to provide cellulose nanocrystals (CNCs). Then, the isolated CNCs from El Diss were used as a reinforcement for a poly(vinyl alcohol-co-ethylene) matrix (EVOH) in the absence and in the presence of borax which was added to improve the interactions between the CNCs and the matrix. The extracted CNCs from both El Diss (CNCD) and El Retma (CNCR) were characterized by Zeta-sizer analysis using dynamic light scattering (DLS), Fourier transform infrared spectroscopy (FTIR), thermogravimetric analysis (TGA), and scanning electronic microscopy. Also, untreated EVOH/CNCD nanocomposites and borax-treated EVOH/CNCD/BOR have been characterized using FTIR, differential scanning calorimetry (DSC) analyses, and by the study of their water absorption behavior. The DLS analysis provided the transversal length of the particles and showed that the surface of the obtained CNCs is negatively charged due to the presence of sulfated ions. Also, FTIR results confirmed the elimination of extra cellulosic substances, whereas TGA proved that the degradation of CNCs occurs at relatively lower temperatures compared with the neat fibers. The incorporation of borax to EVOH/CNCD nanocomposites showed its efficiency in improving the interactions at the interface between EVOH and the CNCD, which significantly affected the material’s thermal properties as concluded from DSC results and their water absorption behavior.
Introduction
The development of new materials that combine attractive performances, environmental compatibility, and renewable source actually consists of a potential sustainable alternative to many petroleum-based materials. 1 The rising of the scientific interest in developing bio-based materials and composites using renewable resources is indicated by the steady increase of the number of publications in this topic and the huge advances in improving the properties of biomaterials and biocomposites. 2
The use of natural fibers as fillers for polymers continues to receive a privileged interest due to the various improvements concluded on the mechanical properties, among other properties. 3 Indeed, the main reasons that have extended the use of vegetable fibers as a principal component of biocomposites are their vast availability, less energy consumption, low-cost production, biodegradability, low density, and high specific strength. 4
Additionally, the idea of extraction of nanocrystals from natural fibers to produce bio-based nanofillers and promote bio-nanocomposites emerged concomitantly with the development of nanocomposites reinforced with nanominerals. 5 The aptitude of cellulose to provide nanocrystals (CNC) is conferred by its semicrystalline structure. 6
Nanocrystals derived from cellulose hold a great promise for an ideal strategy for sustainable development. 7 They present exceptional characteristics as a high-elastic modulus that is of the order of 100–130 GPa and dependent on the original fiber source as well as a specific surface of several hundred m2 g−1 that can be exploited to reinforce polymeric matrices. 8
CNCs have been extracted from several lignocellulosic sources such as wood, pulps, bagasse, cotton, and recycled paper. Also, numerous approaches for producing CNCs in terms of isolation efficiency and environmental impact have been proposed. 9 In this context, acid hydrolysis, alkaline and oxidative degradation processes were found to be the most effective routes to extract highly crystalline cellulosic microfibrils. However, the most commonly used process is acid hydrolysis, which involves the removal of amorphous components covering the cellulose fibrils using a strong acid, which in turn results in individual crystallites forming relatively stable suspensions. 10,11
The use of CNCs as a reinforcing phase in a wide variety of polymers matrices has been reported. 12 Favier et al. 5 reported an earlier mechanical enhancement upon the incorporation of tunicate whiskers into a poly(styrene-co-butyl acrylate) latex. This was explained by the formation of a percolating whiskers network in the polymer matrix, in which stress transfer among the whiskers is facilitated by hydrogen bonding. Geng et al. 13 dispersed CNCs in a poly(vinyl acetate) (PVAc) matrix and found that the in situ polymerized PVAc/CNC composite presented a better dispersion than that showed by the composite produced using mechanical mixing. The investigation of the mechanical and thermomechanical properties revealed that the in situ polymerized nanocomposite with 10 wt% of CNCs exhibited higher strength and storage modulus compared with the mixed composite with the same CNC concentration. Also, Hammiche et al. 14 used CNCs extracted from Alfa to reinforce a poly(vinyl chloride) matrix and obtained noticeable improvements in thermal and thermomechanical performances.
Besides, CNCs are very difficult to disperse in polymers and tend to agglomerate due to their limited stability in aqueous suspension resulting from the high tendency of cellulose nanofibrils to form hydrogen bonds between each other. 15 Although many methods have been conducted to remedy this problem and improve the dispersion of CNCs in polymer matrices, surface chemical modification was found to be the most efficient. 13
The aim of this work is to explore the possibility and feasibility of efficiently extracting CNCs from two local plants, El Diss and El Retma, thus providing potential candidates for the cost-effective production of CNCs. Afterward, CNCs extracted from El Diss (CNCD) were incorporated into a poly(vinyl alcohol-co-ethylene) (EVOH) matrix to produce biodegradable materials presenting a better water resistance with respect to composites prepared from natural fibers. To avoid agglomeration of CNCD and ensure their optimal dispersion, sodium tetraborate was added to induce EVOH cross-linking, which could significantly restrict CNCD movements and prevent their agglomeration during the nanocomposites processing. Additionally, EVOH cross-linking is expected to notably enhance the material’s thermal stability and mediate the high hydrophilic character of the used matrix. After characterizing the microstructure and dimension of the extracted CNCs, the thermal properties and water uptake aptitude of the EVOH/CNCD composites were also studied.
Experimental
Materials
CNCs were extracted from two plants, El Diss and El Retma, which are abundant in the Sétif (Algeria) region. The reagents used for the extraction process, such as acetone (CH3COCH3), sodium hydroxide (NaOH), acetic acid (CH3CO2 H), sodium chlorite (NaClO2), and sulfuric acid (H2SO4), were purchased from Sigma-Aldrich (St. Louis, Missouri, United States). Ethanol (CH3CH2OH) was purchased from Biochem Chemopharma (Montreal, Quebec, Canada). EVOH used for the preparation of EVOH/CNCD nanocomposites has the following characteristics: ethylene concentration, 44%; density, 1.140; and melting point, 165°C; EVOH was also provided by Sigma-Aldrich. Dimethylsulfoxide (DMSO) and sodium tetraborate decahydrate (borax; Na2B4O7·10H2O) used, respectively, for the preparation and treatment of composites were supplied by Sigma-Aldrich.
CNC’s extraction from El Diss and El Retma fibers
El Diss and El Retma nanocrystals (CNCs) were prepared by using the method reported earlier by Hammiche et al. 14 This protocol involves three essential steps, which could be described as follows. The first step involves a pretreatment that isolates cellulose by complete or partial elimination of lignin and hemicellulose. A chemical treatment with a strong acid (H2SO4) is performed to hydrolyze the amorphous parts in cellulose. At last, the obtained CNCs are dispersed in water to form a stable solution.
Pretreatment
After being washed, dried, and cut, the neat fibers were powdered in a coffee grinder and then sieved to get an average uniform particle size around 100 µm. The powder was treated with acetone/ethanol (38/62; V/V) mixture for 24 h.
Alkali treatment
To remove lignin and hemicellulose, the acetone/ethanol pretreated fibers were subjected to an alkaline treatment carried out five times for 2 h, under magnetic stirring, in a 3% NaOH solution kept in a water bath maintained at a temperature of 80°C. A wash with distilled water was performed after each treatment to remove the residual alkaline solution.
Bleaching of the alkali-treated fibers
The bleaching treatment was performed with a sodium chlorite/glacial acetic acid mixture consisting of equal parts of an acetate buffer (27 g of NaOH and 75 mL of glacial acetic acid and diluted to 1 L of distilled water) and aqueous chlorite (17 g of NaClO2 in 1 L of distilled water) at 120°C for 2 h under magnetic stirring. This procedure was repeated four times.
Acid hydrolysis
The hydrolysis of the bleached fibers was carried out with a 65% sulfuric acid solution at 50°C in a water bath, for about 1 h, under stirring to remove amorphous regions. Immediately after the acid hydrolysis, the suspension was diluted 10-fold with distilled water to quench the reaction. Successive washings in a centrifuge Universal 16-Hettich (Tuttlingen, Germany) at 5000 rpm for 30 min were then performed. To obtain a stable solution and a better dispersion of CNCs, the CNC solution (concentration: ∼0.4% in mass) was treated with ultrasound by a 20 kHz frequency ultrasonic generator for 120 min in water bath using a Vibra-Cell™ Ultrasonic Processor (130 W). Vibra-Cell™ VC 75185, Thermo Fisher Scientific Inc., Waltham, MA, USA.
Preparation of EVOH/CNC formulations
Casting/evaporation method was used to prepare 1% and 3% untreated (EVOH/CNCD) nanocomposites and borax-treated (EVOH/CNCD/BOR (borax)) nanocomposite films.
Untreated EVOH/CNCD nanocomposite films
A mixture of EVOH and DMSO at 10% (W/V) was placed in a water bath at 140°C under continuous stirring for 30 min. In parallel, the CNCD (0.4%) were sonicated for 2 h, then added to the EVOH solution and stirred until homogenization followed by sonication for 5 min. The mixture was poured into a previously heated dish and then dried in an oven at 60°C until water and DMSO were evaporated. Afterward, the obtained films were immersed in a water bath for 24 h to remove the DMSO traces and finally dried at 60°C.
Treated EVOH/CNCD nanocomposite films
EVOH/CNCD/BOR formulations were prepared according to the same procedure described above. For the dispersion of CNCD, 10 ml of 4% borax solution was added to the polymer under stirring before being poured into a previously heated dish.
Characterization techniques
Characterization of CNCs
Zeta-sizer analysis: The particle size distribution of CNC samples was estimated by dynamic light scattering (DLS) using a Zeta-sizer nano ZS instrument (Malvern, UK). The dried CNC samples were redispersed in distilled water and then sonicated for few minutes before analysis.
Zeta potential: Surface charges of both the CNCs have been evaluated using zeta potential, which measures the mobility distribution of dispersion of charged particles as they are subjected to an electric field. 12 The analysis was done using DLS (Zeta-sizer nano ZS instrument).
Fourier transform infrared spectroscopy: The chemical composition of raw fibers and dried powders of CNCs was studied using Fourier transform infrared spectroscopy (FTIR). The samples were analyzed with KBr pellets using a Vertex 70v FTIR apparatus (Bruker Corporation, Billerica, MA, USA). FTIR spectra were recorded in absorbance mode, between 4000 cm−1 and 400 cm−1 and at a resolution of 4 cm−1.
Thermogravimetric analysis: The thermal behavior of the raw fibers and dried CNC samples was studied in a temperature interval ranging between 20°C and 700°C, at a heating rate of 10°C min−1, in nitrogen environment (purge rate details: balance chamber flow rate = 30 cm3 min−1 and furnace flow rate = 150 cm3 min−1) using a Mettler Toledo thermogravimetric analysis (TGA)/differential scanning calorimetry (DSC) apparatus (Columbus, Ohio, USA).
Scanning electronic microscopy: The morphological characterization of raw fibers and CNC samples was done with a scanning electronic microscope (SEM) using a JSM-6460 apparatus (JEOL Inc., MA, USA).
Characterization methods for EVOH/CNCD nanocomposites
FTIR spectroscopy: The molecular interactions between the matrix and CNCD were investigated by FTIR spectroscopy. The analysis of the EVOH/CNCD nanocomposite films was performed using a Vertex 70v spectrometer, in absorption mode, in the range between 4000 cm−1 and 400 cm−1 at a nominal resolution of 4 cm−1.
Differential scanning calorimetry: The thermal characteristics of the nanocomposites before and after the treatment were determined using a Mettler Toledo DSC 3 calorimeter under nitrogen flow and at a heating rate of 10°C min−1. For the analysis, the following heating program has been carried out: heating from 25°C to 200°C, annealing for 2 min at 200°C, cooling from 200° to 0°C, a second annealing for 2 min at 0°C, finally a heating from 0°C to 200°C. From the DSC thermograms giving the variations of the heat flow versus temperature, the glass transition, crystallization, and melting temperatures, respectively, (T g), (T c), and (T m) as well as the crystallization and melting enthalpies, respectively, ΔH c and ΔH m, have been determined. The crystallinity degree (χ c) for each formulation was calculated according to the following equation:

ω is the EVOH mass fraction of the composite. The considered melting enthalpy for a 100% crystalline EVOH (
Water absorption measurements: Films were cut into samples of approximately 1 × 1 cm2, which were dried in a vacuum oven at 100°C for 24 h. The initial weights of film samples were evaluated using a Mettler Toledo analytical balance (Columbus, Ohio, USA) with a resolution of 0.0001 g. The samples were then immersed in a closed vessel containing water at room temperature for 72 h. Water uptake measurements were recorded at 24 h intervals and then calculated according to the following equation:

where mt is the mass of the sample at time t and m 0 the mass of the sample before immersion into water. 3
Results and discussion
Characterization of CNCD and CNCR
CNC sample size characterization
Figure 1(a) and 1(b) presents the size distribution of CNC samples extracted from El Diss and El Retma fibers as determined from DLS, respectively. The figures show most of the particle sizes were between 60 nm and 190 nm and 220 nm and 600 nm for CNCD and CNCR, respectively. About 60% of CNCD reveal a diameter between 70 nm and 170 nm, 20% are less than 100 nm, and 20% are between 220 nm and 460 nm. However, CNCR nanoparticles show a larger distribution (>200 nm), where 37% are in the range of 200–300 nm, 60% are between 300 nm and 500 nm, and 6% have a diameter of 530 nm. The discrepancies in particle sizes between the two types of the fibers suggest that El Diss fibers are more susceptible to the performed extraction process, in particular the acid treatment, compared with those derived from El Retma and confirm that particle sizes of CNCs principally depend on their source. 17 The large distribution of both CNCD and CNCR may be explained by the eventual existence of nanoparticles aggregates in water, as has been observed by Tang et al. 7 when extracting CNCs using phosphoric acid. However, Hammiche et al. 14 suggested that the large size distribution obtained for CNC derived from Alfa fibers is due to their big aspect ratio and that the observed dimensions are influenced by the orientation of CNCs in the solution. Additionally, it is important to point out that the DLS technique probes the particle size based on a spherical model. So when CNCs are not agglomerated into a sphere-like morphology, the evaluated dimensions would still be at least the size of the CNC lengths.

Diameter size distribution of CNCs extracted from (a) El Diss and (b) El Retma fibers.
Morphological analysis
Figure 2(a) and 2(b) shows the morphology of El Diss and El Retma raw fibers with different scales, respectively, as determined by SEM. The images reveal that the fibers present different shapes and irregular size distribution. The fiber width varies from 16 µm to 85 µm for El Diss fibers and from 16 µm to 66 µm for El Retma ones. The particles are bonded together due to hydrogen interactions. 18

SEM micrographs of (a) El Diss and (b) El Retma raw fibers.
SEM images of dry CNCD and CNCR are given in Figure 3(a) and 3(b), respectively. The figures exhibit CNC aggregates resulting from the self-assembled fractal structures due to the occurrence of strong hydrogen bonds. These interactions are particularly favored due to the hydroxyl groups existing at the surface of the cellulose fibrils and are highly dependent on the cellulose source and extraction methodology. 10 After CNC drying, the interactions increase, thereby making the rupture of aggregates difficult even after redispersion into water. Such observations agree well with the DLS findings which pointed out the presence of CNCs with variable sizes of CNCD and CNCR in the suspensions in water and with the results that have already been reported by many other authors. 19,20

SEM micrographs of dried (a) CNCD and (b) CNCR.
Zeta potential
Zeta potential results have shown that both CNCD and CNCR manifest negative surface charges of about −39.8 mV and −15 mV, respectively, due to the substitution of the hydroxyl groups present in the cellulosic material by the sulfate ester groups brought by the sulfuric acid used during the hydrolysis step. The presence of these electric charges induces an electrostatic repulsion, which would contribute to the prevention of particle aggregates and thus helps to generate a stable solution of CNC. 21 In this context, it has been proposed that to obtain colloidal stability, the value of the zeta potential must exceed −30 mV, whereas a value of −15 mV announces the beginning of particle agglomeration. 22
The obtained values show that the zeta potential value of CNCD is higher than that evaluated for CNCR. This is an expectable result because CNCD presents more hydroxyl groups than CNCR as revealed by their lower particle sizes. This implies that CNCD presents a larger number of hydroxyl groups that could be esterified by the sulfate ions. On the contrary, CNCR reveals a limit value for a stable solution. These results fit well with the diameter size distribution which indicated for CNCD a narrower particle size distribution profile than that obtained for CNCR. This suggests that during acid hydrolysis, El Diss microfibrils expose more hydroxyl groups to esterification by sulfate ions than El Retma ones. However, El Retma fibrils are less susceptible to esterification probably due to their pronounced tendency to agglomeration. CNCs’ characteristics obtained after hydrolysis vary according to the imposed conditions; for example, hydrolysis time and acid-to-pulp weight ratio which affect the particles’ dimensions and hydrolysis temperature which could cause dehydration and/or carbonization of the final product. 23 In this case, we could propose that hydrolysis conditions were not enough to induce a sufficient esterification rate for El Retma fibers hydroxyl groups by sulfate ions of the treating agent.
Structure analysis of CNCD and CNCR
Figure 4(a) depicts the FTIR spectra of El Diss and El Retma raw fibers. The confirmation of the cellulosic structure of the analyzed samples is provided by the appearance of the main functions that can be enumerated as follows. The large band observed in the range comprised between 3650 cm−1 and 3000 cm−1 of all the spectra is attributed to the stretching vibrations of hydroxyl groups contained in cellulose and lignin. 24 The small bands detected around 2875 cm−1 and 1500 cm−1 are characteristic of C-H stretching vibrations of lignin. The presence of hemicellulose and pectin is confirmed by the characteristic band between 1730 cm−1 and 1745 cm−1 attributed to the stretching of carbonyls of the acetyl groups present in these substances. The O-H bending vibration of adsorbed water appears around 1630 cm−1 and is due to the hydrophilic character of the cellulosic fibers. 25,26 The band at 1428 cm−1 is attributed to the bending of –CH2 groups. 27 The stretching vibrations of C=C bonds in the aromatic rings of lignin are observed in the range of 1500–1600 cm−1. 18 The skeletal vibrations involving C−O stretching appear around 1110 cm−1 and 1060 cm−1. 28 The band in the region of 1161 cm−1 is due to asymmetric stretching of C–O–C of glucose ring in cellulose structure. 29

FTIR spectra of (a) El Diss and El Retma raw fibers and (b) CNCD and CNCR.
The FTIR spectra of CNCD and CNCR are represented in Figure 4(b). The characteristic absorption bands which are displayed by the FTIR spectra of the two samples confirm their cellulosic structure. Also, it is found that acid hydrolysis does not damage the cellulose microstructure of the microfibrils. After hydrolysis treatment, the extraction of CNCs is evidenced by the small band at 888 cm−1 attributed to the antisymmetric out-of-plane stretching of glucose ring in cellulose appearing for CNCR. Additionally, new bands due to the deformation vibrations of CNC groups are detected around 1151 cm−1 and 1193 cm−1 for CNCD and at 1173 cm−1 for CNCR. These confirm that the obtained CNCs have the structure of cellulose I. 27 In this context, Fortunati et al. 27 reported that the weak signal around 800 cm−1 confirms the existence of CNCs and their monomeric units after hydrolysis treatment. Also, the presence of signals at 1426, 1173, 1099, and 896 cm−1 indicates that the CNCs present primarily the cellulose I microstructure. Moreover, the CNCD and CNCR FTIR spectra show that the large band observed between 3000 cm−1 and 3700 cm−1 and corresponding to –OH stretching vibration is more prominent compared to raw fibers. This indicates a higher cellulose content and the formation of a large amount of OH groups due to the decomposition of β-1,4-glucosidic linkage. 25 Furthermore, the reaction of cellulose with sulfuric acid is supported by the appearance of two new bands around 1200 cm−1 which could be assigned to sulfate half-ester groups. 19
On the other hand, it is observed that after treatment with sulfuric acid, the bands assigned to cellulose structure and related to the amount of molecular disorder, particularly those corresponding to –OH groups stretching vibrations and –C–H– stretching vibrations detected at 2920 cm−1 become sharper, thus suggesting the increase in the material crystallinity. Similarly, Tang et al. 7 proposed that the increased absorption intensity may be considered to be related to the increased crystallinity of CNC after hydrolysis and enzymatic treatment. The evaluation of the absorbance ratio at the wavenumbers of 1433 cm−1 and 888 cm−1 is considered as an adequate route for the comparison of CNCD and CNCR crystallinity indexes. 30 Their estimation leads to a value of 1.056360 for CNCR and 1.125287 for CNCD, which implies that the crystallinity index of the CNCD is higher than that of CNCR. Figure 5 shows the reaction pathway of cellulose hydrolysis with sulfuric acid.

Reaction of cellulose acid hydrolysis with sulfuric acid.
Thermal stability assessment by TGA
TGA was performed to compare the raw and acid hydrolyzed cellulose compounds degradation characteristics, as determined from the thermograms reporting the variations of the weight loss (thermogravimetric (TG)) and the derivative of the weight loss versus time (derivative thermogravimetry).
Generally, cellulose degradation processes consist of dehydration, depolymerization, and decomposition of glycosyl units, followed by the formation of a charred residue. 31
Figure 6(a) and (b) gives the weight loss and its derivative versus time, respectively, and shows that El Diss and El Retma raw fibers exhibit three main weight loss regions that correspond to different decomposition temperatures for the various components (hemicellulose, lignin, and cellulose) of the two natural fibers.

(a) TG and (b) DTG curves of El Diss and El Retma raw fibers.
The initial weight losses were due to the evaporation of chemisorbed water and retained moisture on the surfaces of El Diss and El Retma raw fibers. The main degradation stage, occurring between 220°C and 350°C, corresponds to the thermal decomposition of hemicellulose, pectin, and the cleavage of glycosidic linkages of cellulose. 31 The peak that appears around 260°C is due to the decomposition of hemicellulose and is more prominent for El Diss raw fiber. The shoulders observed above 350°C are attributed to lignin decomposition. It has been proposed that lignin begins to decompose at a lower temperature than that of cellulose; but at higher temperatures, lignin is more heat resistant than hemicellulose and cellulose because of its low degradation rate. 14
On the other hand, CNC’s thermal degradation behavior, displayed in Figure 7(a) and 7(b), presents three main decomposition stages corresponding, respectively, to the evaporation of moisture at low temperature (below 100°C), the primary pyrolysis (150–300°C), and finally the slow charring process of the solid residue (after 300°C). 32 Indeed, the first weight loss of CNCD and CNCR is detected between 60°C and 140°C and is attributed to the evaporation of absorbed water. 31 The fraction of evaporated water is more important in the case of El Diss nanocrystals, which corroborates the fact that they contain a higher concentration of –OH groups, as it has been stated from FTIR and Zeta potential results. This also indicates that they are less aggregated and present subsequently a higher specific area than El Retma nanocrystals. The primary pyrolysis of the CNCD and CNCR begins around 195°C and is principally assigned to the decomposition of sulfated amorphous regions. This temperature is much lower than the temperature at which the decomposition of native cellulose occurs, i.e. in the range between 250°C and 300°C. The second pyrolysis stage initiates around 450°C and consists of the breakdown of nonsulfated crystals. 33 The decrease in the degradation parameters of CNCD and CNCR relatively to those of the raw fibers is due to the released sulfuric acid and the substitution of the hydroxyls by sulfate groups via sulfuric acid hydrolysis. This causes a significant decrease in the activation energy of CNC degradation process. 34 In this context, Mao et al. 9 suggested that the sulfate groups decrease noticeably the thermal stability of natural fiber nanocrystals. They proposed that if these groups are protonated, they are susceptible to catalyzing CNC’s decomposition and if they are unprotonated, they can undergo elimination reactions that also decrease CNC’s stability.
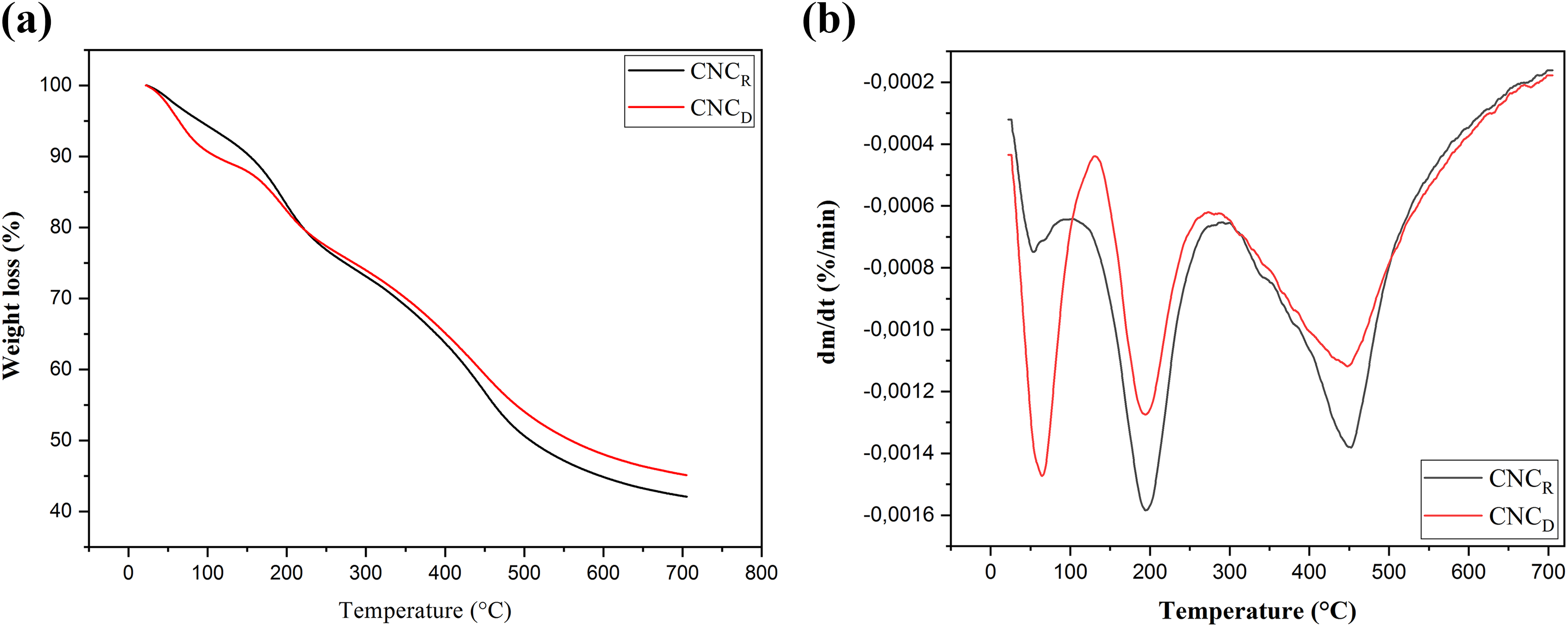
(a) TG and (b) DTG curves of El Diss and El Retma nanocrystals.
To explain the difference between the thermal behaviors of CNCs and cellulose fibers, Lu and Hsieh 19 proposed that the promotion of decomposition–gasification processes into CNCs leads to a decrease in their thermal stability. This is induced by the catalyzing effect exerted by the surface groups and the CNC’s higher surface area meaning a more important exposed surface to heat phonons which results in their higher thermal conductivity. Hence, the smaller crystallized cellulose chain bundles permit a much more phonon scattering in CNCs than in amorphous chains contained in cellulose, which explains their relatively higher thermal conductivity. Also, Mao et al. 9 reported that the early decomposition of sulfonated CNCs is promoted by the increased free volume around the areas in the highly sulfated regions, namely the surface and the chain ends.
Characterization of EVOH/CNCD nanocomposites
Nanocomposites structure analysis
The evolution of the chemical structure for EVOH/CNCD and EVOH/CNCD/BOR nanocomposites before and after treatment with borax was investigated by means of FTIR analysis, and the results are reported in Figure 8.

FTIR spectra of untreated and borax-treated EVOH/CNCD nanocomposites at 1% of CNCD.
The chemical structures of the two nanocomposites are similar and the most characteristic cellulose bands overlap with infrared bands of EVOH. Many characteristic bands of common functional groups to both cellulose and EVOH structure appear in the range 3600–3000 cm−1 and between 3000 cm−1 and 2800 cm−1 and are assigned to the hydroxyl groups and alkyl skeleton stretching vibrations, respectively. 30 The FTIR spectra of 1% borax-treated EVOH/CNCD/BOR films show important structural variations because of the appearance of two additional bands at 955 cm−1 and 1015 cm−1 due to the presence of CNCD and the decrease in the intensity of the hydroxyl groups. Fortunati et al. 35 explained such a decrease in the occurrence of interactions involving the OH groups brought by the CNC nanoparticles and those owing to the EVOH matrix, which is particularly favored in the presence of borax.
Thermal properties of EVOH/CNCD nanocomposites
DSC measurements have been performed to provide further information about the eventual interactions of CNCs and the EVOH matrix and on the effects of borax. Figure 9(a) and (b) presents, respectively, the cooling and second heating DSC thermograms of neat EVOH and untreated and borax-treated EVOH/CNCD nanocomposites.

(a) Cooling and (b) second heating DSC thermograms of EVOH/CNCD and EVOH/CNCD/BOR nanocomposites.
Neat EVOH shows a glass transition temperature around 72°C and a crystallinity that approximates 23%. After the incorporation of CNCD into the untreated nanocomposites, significant variations are noticed on the whole thermal characteristics of the matrix. The glass transition temperature decreases abruptly by 20°C to attain a value of 52°C. Also, the crystallization exotherm area increases and the maximum of the peak appears at a lower temperature, which is 138°C for the untreated nanocomposite containing 1% of CNCD. The crystallization exotherm disappears completely for the nanocomposite loaded with 3% of nanocrystals, which suggests that it has totally crystallized during the cooling step following the processing. As a result of these noticeable changes in the crystallization process, the melting behavior of the untreated nanocomposites shows an important decrease in the melting temperature and an increase in the crystallinity. These results support the fact that the incorporation of CNC induced a substantial plasticizing effect that has contributed to the decrease of the matrix T g and favored the chains’ mobility that has engendered a higher crystallinity and reduced the crystallization temperature. Also, due to this high nucleating effect, the EVOH/CNCD nanocomposite crystals seem to be less perfect and/or smaller than those formed by the neat matrix, which explains the decrease in the melting temperature. Similar results have been reported when studying PLA/CNC nanocomposites and have been justified by the presence of residual solvent in the films after the casting process. 35
EVOH/CNCD/BOR nanocomposites presented a totally different thermal behavior from that exhibited by the untreated nanocomposites, due especially to EVOH cross-linking and CNC’s aggregation. The matrix T g increases to 64°C and 57°C, relatively to the untreated nanocomposites, for the formulations with 1% and 3% of treated CNCD nanocomposites, respectively. This means that the plasticizing effect has been attenuated thanks to the interactions established by the borax at the interface between the nanocrystals and the polymer matrix, which limited the chains’ mobility. 36 The nanocomposite with 3% of CNC presents a lower T g due to the fact that the higher CNC’s concentration into the matrix caused probably more agglomerations and less interactions at the interface. This result fits well with that reported for polyvinyl alcohol (PVA)/sisal nanofibers. 37 Additionally, the crystallization exotherms are significantly reduced and their maximum displaced toward lower temperatures implying that a more important supercooling is necessary for the nanocomposites to undergo crystallinity. Due to the disadvantaged crystallization process for the borax-treated CNCD nanocomposites, the melting enthalpy and the crystallinity values are notably decreased. Also, the EVOH crystals seem to be less perfect and smaller and so they melt at lower temperatures, relatively to those of neat EVOH and untreated EVOH/CNCD nanocomposites. All these results support the fact that the incorporation of borax induced EVOH cross-linking which increased notably the matrix T g and hindered its aptitude to crystallization as confirmed from crystallinity values presented in Table 1, which reports the principal thermal characteristics of neat EVOH and EVOH/CNCD and EVOH/CNCD/BOR nanocomposites.
Thermal characteristics of EVOH/CNCD and EVOH/CNCD/BOR nanocomposites.

EVOH: poly(vinyl alcohol-co-ethylene) matrix; CNCD: CNCs from El Diss; BOR: borax.
Absorption behavior
The behavior of the untreated and borax-treated nanocomposites regarding water absorption is shown in Figure 10. The hydrophilic nature of EVOH allows the absorption of significant amounts of moisture in high-relative humidity conditions. In fact, the polar hydroxyl groups contained in EVOH structure cause its moisture sensitivity because they are not totally self-associated and are partly isolated in the matrix which interacts with water molecules via hydrogen bonding. 38 According to Figure 10(a), EVOH neat matrix presents a mass gain of 8.97%, which is compatible with the results reported earlier by other authors such as Lagaron et al. 39 who observed a weight gain of 9.0%, while Aucejo et al. 40 and Zhang et al. 41 who found values of 13% and 8.4%, respectively, for EVOH films with 32 mol% of ethylene. EVOH water sensitivity was seen to decrease with the increase in ethylene content and degree of crystallinity. The crystallinity increases with vinyl alcohol unit content which restricts chain mobility by intermolecular hydrogen bonding interactions. On the other hand, the increase in the vinyl alcohol unit means increasing hydroxyl groups that will interact with water molecules. 38

Percentage of water absorption for EVOH/CNCD and EVOH/CNCD/BOR at 1% of CNCD after (a) 72 h and (b) 576 h of water uptake.
For EVOH/CNCD nanocomposites, water absorption occurs simultaneously due to the EVOH matrix through the polar sites present in the polymer, and the cellulose whiskers where the –OH groups of the glucose are a potential site for water uptake and at the interface polymer/CNCD. 42 For the untreated sample filled with 1% of CNCD, only a mass gain of 1% over that of a neat EVOH matrix is noticed. The poor water absorption for EVOH/CNCD nanocomposite is due to the fact that crystalline cellulose does not absorb significant amounts of water. This also implies that in this case, the water uptake is related predominantly to the EVOH matrix and EVOH/CNCD interface. 43 Also, the weight gain is not significant, thus suggesting a good adhesion between the two components. Additionally, Silvério et al. 44 reported that the agglomerations of the CNCs dispersed all along the matrix create a barrier against water absorption. Analogously, Saxena 45 agreed that CNCs form a rigid hydrogen-bonded network, which stops the diffusion of water molecules. However, other studies demonstrated an opposite behavior where water absorption increases with the particles’ agglomeration. 46,47
In contrast to EVOH/CNCD nanocomposite, the EVOH/CNCD/BOR one with 1% of CNCD shows higher water absorption compared with neat EVOH and its untreated counterpart. This could be interpreted by the fact that after dissolving borax in water, the association of the two compounds generates tetrahydroborate ions
Furthermore, Figure 10(b) shows that the samples’ weights decrease when the immersion time is prolonged. This behavior could be explained by the dissolution of PVA contained in the matrix. The weight loss is much more significant for the virgin matrix and decreases after the addition of the CNCD. This behavior points out the contribution of the interactions between CNCD and the polymer matrix through their hydroxyl groups in preventing PVA solubilization. Similarly, the material treated with borax exhibited a decrease in weight loss with increasing immersion period, thus corroborating the cross-linking of the EVOH and the improvement of the interactions at the interface CNCD/matrix.
Conclusion
In the present study, CNCs have been extracted from two abundant local plants which are El Diss and El Retma and then characterized. Afterward, the CNCs obtained from El Diss (CNCD) have been incorporated in an EVOH matrix. The characterization of CNCD and CNCR extracted from El Diss and El Retma, respectively, pointed out, from the analysis of nanoparticles size distribution and Zeta potential measurements, the strong influence of the type of native fibers on the final obtained products. Also, FTIR results evidenced the effectiveness of the performed treatments in the elimination of extra cellulosic materials. This engendered CNCs with a lower thermal stability compared with the corresponding native fibers as deduced from TGA analysis which also confirmed the surface esterification of CNCs.
The treatment with borax, used to improve the dispersion of CNCD in the EVOH matrix, induced significant variations in the thermal properties of the matrix, thus stating for a better CNCs dispersion and favorable interactions at the interface between the two components. The highly crystalline structure caused a slight increase in the nanocomposites water absorption aptitude that increased in the presence of borax due to the formation of tetrahydroborate ions which introduce more hydroxyl groups into the nanocomposite. It was concluded that by increasing the immersion time, PVA phase dissolution was prevented because of the interactions coupling the matrix to the CNCD nanoparticles and the cross-linking of EVOH.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.