
Abstract
This article is focused on the preparation and characterization of functional nanosilica incorporated dental resins with better mechanical, cytotoxicity, sorption, and solubility properties. Silica nanoparticles were synthesized via Stöber method and were functionalized with 3-amino-1,2,4-triazole. Dental nanocomposites were produced by embedding the functionalized nanosilica into bisphenol-A-glycidyl methacrylate/triethylene glycol dimethacrylate matrix to form B1–B6 series. This was achieved by mechanical mixing of the monomer (50:50 wt%), filler (10–60 wt%), initiator combination (CQ/EDMAB:0.1:0.4 wt%) and then followed by LED light curing (wavelength: 450–500 nm, power density:1000 mW cm−2) for 60 s. Fourier transform infrared spectroscopy, scanning electron microscopy, and thermogravimetric analysis techniques were used for characterization of the materials. Cytotoxicity tests were performed to evaluate cell viability and mechanical tests were done to check mechanical strength and stability of the materials. The mean sorption and solubility values of the materials were measured by making a series of experiments on different composite formulations.
Introduction
Polymer-based resins have been used for decades in dental treatment technology. 1,2 Although these materials have been employed as promising candidates, these resins are facing serious problems because of restoration failure, inadequate mechanical strength, poor wear resistance, and high volumetric shrinkage during polymerization. 3 The volumetric shrinkage may lead to a large incidence of secondary dental caries and a high potential for discoloration and marginal leakage. 4 These problems can profoundly influence the restoration performance of dental composites. The long-term performance of composite materials in clinical dentistry is one of the important directions that needed to resolve the drawbacks associated. 5
In order to have better dental composite resins that provide better clinical performance, several attempts have been made on the polymer matrix or volume of the fillers. 6 Physical properties of the dental composites can be greatly influenced by the particle size and filler volume, where the latter is related to size, silanization, and loading. 7 -9 It was demonstrated that the polymerization shrinkage decreased with increasing the volume fraction of the filler in the polymer matrix. In the meanwhile, the hardness, compressive strength, elastic modulus, and flexural strength of the composite materials are improved. 7 The properties of the polymer matrix can also be enhanced by means of focusing on the alternative monomer production. 10 -12
Modification of the materials, which included filler content optimization, 13 packing, 14 and development of hybrid additives, 15 has been recognized as an alternative and effective way of performance improvement of polymer composite resins.
Recently, incorporation of nanoparticles (NPs) as fillers offered several advantages, such as reducing polymerization shrinkage, improving mechanical properties, and rate of wear. 16 -19 The NPs have a bigger surface area and include surface functional groups, which are the main reason for the improvement of the material. 20 Although the addition of NPs in the resin has shown a number of advantages, further progress is needed to obtain long-term restoration purpose in clinical therapy. 21,22
Azole scaffolds are considered as the most viable structure to produce effective antimicrobial agents. These materials have shown significant antibacterial activities on methicilin-resistant strains of Staphylococcus aureaus. Pharmacokinetic properties of azoles illustrated that they have weakness in their safety and potency. The structure of the available effective azole drugs was modified to enhance their antimicrobial potency and selectivity. 23
The objective of this work is to search physicochemical and biological properties of azole linked nanosilica incorporated dental nanocomposite resins. The SiO2 NPs were produced using Stöber method and their surfaces were modified with 3-amino-1,2,4-triazole to form functional silica nanoparticles (f-SiO2 NPs). Dental nanocomposites B1–B6 were prepared with the addition of different weight percentages of f-SiO2 NPs fillers (10–60 wt%) into a blend of bisphenol-A glycidyl methacrylate/triethylene glycol dimethacrylate (BisGMA/TEGDMA) using a photoinitiator system. The functional groups were characterized by Fourier transform infrared spectroscopy (FTIR) and the surface morphology of the prepared nanocomposites was analyzed by scanning electron microscopy (SEM). Thermogravimetric analysis (TGA) was employed to study the thermal stability of the final products. Cytotoxicity, mechanical, sorption, and solubility properties of the materials were analyzed.
Experimental
Materials
The materials and chemicals such as dimethylformamide (DMF, 99.8% anhydrous), BisGMA (>98%), camphorquinone (Mw = 166.22 g mol−1 97%), TEGDMA (99%), tetrahydrofuran (≥99.9%), ethanol (≥99.8% (GC)), tetraethyl orthosilicate (99.9% trace metals basis), ammonium hydroxide (NH4OH, 28.0–30.0% NH3 basis), ethyl 4-(dimethylamino)benzoate (EDMAB) (≥99%), and 3-amino-1,2,4-triazole (≥95% (TLC)) were obtained from Sigma-Aldrich (Taufkirchen, Germany).
3-Amino-1,2,4-triazole modified SiO2 NPs
The synthesis of SiO2 NPs was carried out by sol–gel reaction using Stöber method. 24 -26 The reaction of silica with epichlorohydrin was carried out with a similar procedure according to previous literature. 27 The epoxy functional nanosilica was collected and washed at least four times with 1:4 ratio of excess ethanol/water mixture. The washing step was applied to remove the unreacted impurities from the final product. Finally, the synthesized material was received in wet form for further reaction.
Functional nanosilica were synthesized by the reaction of 3-amino-1,2,4-triazole with epoxy-silica NPs. The epoxy-silica was dispersed in DMF (10 mL) and excess of 3-amino-1,2,4-triazole was dissolved in the mixture and heated at a temperature of 80°C. The solution was stirred under a nitrogen environment for about 24 h. A solid mixture was filtered and washed with 1:4 ratio of excess ethanol/water mixture (four times) and then dried in an oven at 80°C.
Preparation of composite resins
Dental nanocomposite was synthesized with different weight percentages (10–60 wt%) of the 3-amino-1,2,4-triazole functional silica (f-SiO2). Firstly, the monomers BisGMA and TEGDMA (50:50 wt%) were blended and homogenized at 50 C. Then different weight percentages of functional nanosilica were mixed (hand spatulation) for about 25 min until getting a transparent mixture. This process was followed by the addition of an initiator/coinitiator system that is made up of CQ (0.1 wt %) and EDMAB (0.4 wt%). The viscous materials were placed in Teflon mold and LED light source with wavelength: 450–500 nm and power density: 1000 mW cm−2 was used for 60 s to irradiate the mixture. The specimens are labeled according to the weight percentages (10, 20, 30, 40, 50, and 60 wt%) of the f-SiO2 NPs in the polymer composite so-called B1, B2, B3, B4, B5, and B6 (see Table 1).
f-SiO2 percentages of different resin composites series B.
Characterization techniques
The FTIR spectrum was recorded in the range of 4000–400 cm−1 and a resolution of 4 cm−1 with Bruker Alpha-P ATR system. The samples were dried under vacuum prior to analysis. Thermal stabilities of the functionalized fillers and composites were examined by TGA using Perkin Elmer STA 6000. The materials (∼5 mg) were scanned from room temperature to 700°C at a rate of 10°C/min under an N2 atmosphere.
The universal testing machine was used for mechanical properties of the disc-shaped test specimens with 5 mm diameter and 3 mm thickness. The rate of the crosshead was 0.1 mm min−1. Stress–strain (%) curves were recorded to measure mechanical parameters: compression strength or maximum stress, Young’s modulus (E), and strain at break or failure.
Surface morphology of the nanocomposites was analyzed by SEM Philips XL30S-FEG (Nederland). The surface of the composite materials was coated with a conductive layer.
Solubility and sorption tests were performed to analyze water solubility (S) and water sorption (A) of the dental composites according to Oysaed and Ruyter 12 formula. 28 The A and S were formulated as A = m 1−m 2/v and S = mº−m 2/v, where m 1, m 2, and mº are the sample weight before immersion, after immersion, and desiccation, respectively. v is the volume of the specimen in cubic millimeters.
For cytotoxicity studies, discs (3 mm thickness and 5 mm diameter) of the dental composites with uniform wells were handled together with Teflon control. Ultraviolet light was used to sterilize the discs under the tissue culture hood for 20 min. Then, discs were transferred into a 15-mL conical tube. Sequentially, the specimens were washed using 70% ethanol/water mixture and left to dry in the hood for 5 min. Afterward, each disc was placed in a well of a 24-well plate and preincubated in 1 mL of Dulbecco’s modified Eagle medium (DMEM) for 15 min.
HepG2 cells (ATCC) were seeded at 1 × 105 cells/well in a 24-well plate comprising DMEM with 10% FBS, 100 U mL−1 penicillin, 4.5 g L−1 glucose, 100 µg mL−1 streptomycin, and 2 mM,
Results and discussion
FTIR analysis
Figure 1(a) and (b) shows the FTIR spectra of nano-SiO2, f-SiO2 and dental polymer composite resins, respectively. Figure 1(a) shows absorption peak appeared at 3334 cm−1 that is assigned to the O–H stretching vibration and absorption at 1633 cm−1 due to O–H vibration. The broad peak at 1064 cm−1 can be attributed to the Si–O–Si formation. 28 -30 The peaks at 1220 and 1083 cm−1 are assigned to the Si–O stretching. The absorption at the middle of 954 cm−1 is due to the stretching of Si–O and Si–O nonbridging oxide. The band at around 795 cm−1 is attributed to the stretching symmetry of Si–O–Si mode. The least absorption bands are at 550 and 470 cm−1 belong to the Si–O–Si plane perpendicular rocking motions of the bridging oxygen adjacent to the two Si atoms that formed trisiloxane rings. 30 -32 The f-SiO2 has absorption peaks at 1420, 1520, and 1670 cm−1 belonging to azolic ring.

FTIR spectra of nano-SiO2 and functionalized silica, f-SiO2: (a) dental composite specimens, B1–B6 and (b) uncured specimen (bottom spectrum was drawn in dark blue).
The characteristic group peaks of polymer composite resins as well as uncured sample are shown in Figure 1(b). Peaks at 2963 cm−1 to 2850 cm−1 correspond to –CH3 and –CH2 groups (asymmetric and symmetric stretching, strong and sharp) of the polymer. It is clear that the peak is centered at 3479 cm−1 due to C–OH groups of BisGMA, while the 3050–3038 cm−1 small peaks are due to the =C–H bonds (or the aromatic H). The strong peak at 1716 cm−1 is due to C=O group of all methacrylates while the medium, narrow peak at 1636 cm−1 is due to stretch vibration of C=C. The peaks at 1170 cm−1 are attributed to C–O bonds. An absorbance at 1610 cm−1 belongs to the aromatic C=C bonds of BisGMA composite matrix. 33
SEM analysis
Surface morphologies of series B were investigated by SEM, as shown in Figure 2. The pictures of nanocomposite series B2–B6 were obtained after coating the specimens with a conductive layer. It is clear that B2 has a slightly rough surface and the fillers dispersed in the matrix resulting in no phase segregation. In addition, both B3 and B4 have a rough surface and the dispersion of the NPs is homogeneous. Moreover, the surface roughness of B5 is higher than B6 and f-SiO2 NPs are dispersed quite well so that aggregation was observed even at higher additive contents. Consequently, this homogeneous distribution of the nanoadditives significantly improved mechanical properties of the nanocomposites.

SEM, surface images of nanocomposite series, B2–B6.
Thermal analysis
TGA curve representing the composites in series B is shown in Figure 3. The matrix of polymer for the dental resins, investigated previously, gives a 3-D cross-linked structure. The structure is highly resistant to thermal decomposition in an inert atmosphere. A great deal of energy is required to break the polymer bond. 33 The decomposition of the organic matrix and azoles occurs when the temperature is below 400°C. However, when the temperature is above 400°C, the loss in mass is due to inorganic phase. Decomposition behavior of monomers (BisGMA and TEGDMA) is affected by their chemical structure considerably. 34

TGA thermograms of unfilled (B0) and nanocomposite series, B1–B6.
Sorption and solubility analysis
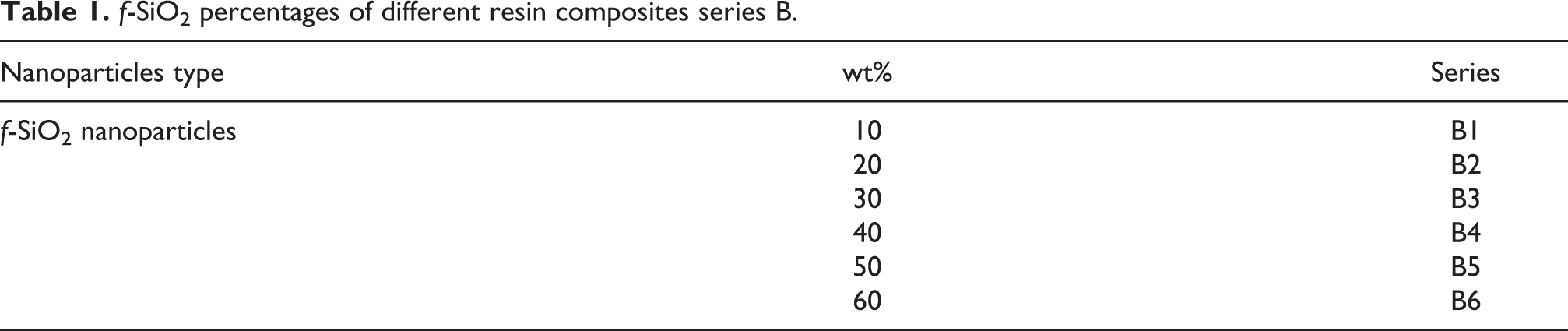
In dental composites, the solubility and water sorption are important properties to assess the debonding of the restoration, recurrent decay, and durability. The results of B series composites indicated that, as the functional NP amount increased, both solubility and sorption properties increased, as presented in Figure 4 and Table 2. It is clear that the water sorption value of composite B1 is 58 µg mm−3, which is close to the required value of ISO 4049. 35 Then, sorption values increased as the filler content increased, whereas solubility values exhibited a similar trend at lower contents up to 40% and then increased. These data implied that lower solvent sorption and solubility for B1--B3 samples, whereas these values increased with the additive content for B5–B6 nanocomposites. These values were received after storage in water in an oven at 37°C for 3 weeks.
Solubility and sorption of the composites B series in water.
Solubility and sorption values of B1–B6.

Cytotoxicity test
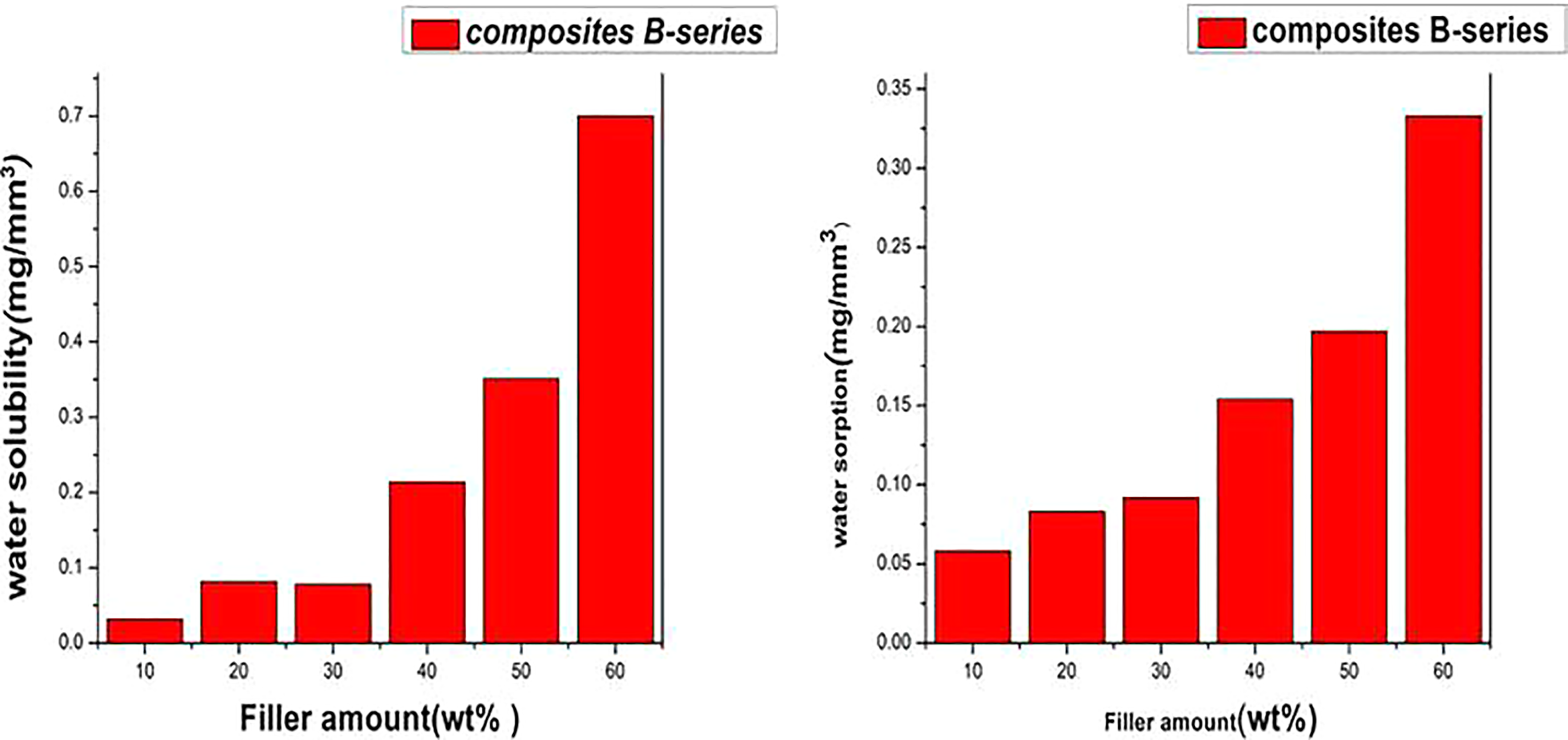
For cytotoxicity studies, the dental composites in the form of discs (3 mm × 5 mm) were produced using a Teflon-based mold with uniform wells. The control sample was a Teflon made disc with almost the same size and shape. Cytotoxicity potential of the different dental composites and control sample was investigated in vitro. Then, it was incubated with HepG2 cells. The assessment of cellular viability was done using the MTT assay. All the details of the sample preparation and test details were described in “Characterization techniques” section. As shown in Figure 5, B1 is less toxic, B2–B4 are moderately toxic, and B5 and B6 are highly toxic, compared to the Teflon-made control. Consequently, the toxicity of the final composites increased with increasing the functional silica content in the composite.
Cell viability mean and SD plots of nanocomposite B series.
Mechanical analysis
Mechanical properties of the B series were investigated over disk specimens with 5 mm diameter and 3 mm thickness via Young’s modulus (E), compression strength, and maximum stress and strain at break (failure). As observed in Figure 6 and Table 3, the first data was the blank sample (resin), where there is no nanoadditive in the material. The results illustrated that the highest compression strength mean value was seen in B1 (303.5 ± 58.7 MPa). As for the strain at break, B5 (6.9 ± 0.78 GPa) showed the highest value. However, the highest elastic modulus was obtained in composite B3 (5.66365 ± 0.42 GPa).

Plot of mean and SD values for (a) compression strength, (b) strain at break, and (c) elastic modulus for composite B and blank.
Mean and standard deviation values of compression strength, strain at break, and elastic modulus for blank and B composites series.
The dental composites with hybrid fillers have elastic modulus and the compression strength values in the range of 5–10 GPa and 150–250 MPa, respectively. 36 -40 The elastic modulus values of prepared composites B1 and B3 are within the reported range for the hybrid fillers. In addition, the mean compression strength of B1 and B2 shows a considerable improvement when compared to dental composites that were made with hybrid fillers.
Conclusions
Silica NPs were successfully synthesized using Stöber method and functionalized with 3-amino-1,2,4-triazole. The functional silica was incorporated into BisGMA/TEGDMA with varying weight percentages. FTIR was carried out to confirm the synthesis of the materials. Composites B1–B6, representing different weight percentages of additives, were investigated by SEM, TGA, sorption and solubility analysis, cytotoxicity test, and mechanical analysis. SEM results confirmed a well dispersion functional nanosilica in the polymer matrix. TGA illustrated that decomposition of the composites starts at around 200°C. The lowest water sorption (0.058 mg mm−3) and solubility (0.032 mg mm−3) were obtained in 10 wt%. Also, cytotoxicity studies on composites B series revealed that B1, B2, and B3 are moderately toxic and B4, B5, and B6 are very toxic compared to the Teflon-made controls. The highest value of compression strength for B1 was 303.5 ± 58.7 MPa and elastic modulus for B3 was 5.37 ± 1.1 GPa. While B5 has the highest mean strain at break value of 6.9 ± 0.78 GPa. Consequently, incorporation of surface modified silica NPs in the polymer resin showed a considerable increase in the mechanical, sorption, solubility, and cytotoxic properties for B1–B3 series. These composites could be suggested for clinical applications.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.