
Abstract
Behavior genetics is the study of the relationship between genetic variation and psychological traits. Turkheimer (2000) proposed “Three Laws of Behavior Genetics” based on empirical regularities observed in studies of twins and other kinships. On the basis of molecular studies that have measured DNA variation directly, we propose a Fourth Law of Behavior Genetics: “A typical human behavioral trait is associated with very many genetic variants, each of which accounts for a very small percentage of the behavioral variability.” This law explains several consistent patterns in the results of gene-discovery studies, including the failure of candidate-gene studies to robustly replicate, the need for genome-wide association studies (and why such studies have a much stronger replication record), and the crucial importance of extremely large samples in these endeavors. We review the evidence in favor of the Fourth Law and discuss its implications for the design and interpretation of gene-behavior research.
Keywords
Behavior genetics is the study of the manner in which genetic variation affects psychological phenotypes (traits), including cognitive abilities, personality, mental illness, and social attitudes. In a seminal article published in this journal, Turkheimer (2000) noted three robust empirical regularities that had by then emerged from the literature on behavior genetics. He dubbed these regularities the “Three Laws of Behavior Genetics.” They are:
1. All human behavioral traits are heritable. [That is, they are affected to some degree by genetic variation.]
2. The effect of being raised in the same family is smaller than the effect of genes.
3. A substantial portion of the variation in complex human behavioral traits is not accounted for by the effects of genes or families.
These observations surprised many outsiders to the field of behavior genetics at the time, yet they remain an accurate broad-brush summary of the empirical evidence 15 years later, as shown by a recent meta-analysis of virtually all twin studies ever conducted (Polderman et al., 2015). Indeed, they have attained the status of “null hypotheses”—the most reasonable a priori expectations to hold in the absence of contrary evidence (Turkheimer, Pettersson, & Horn, 2014).
The original Three Laws summarized results from studies of twins, adoptees, and other kinships. These research designs have many valuable uses, but they cannot discover particular genomic regions or specific variants that are causally responsible for downstream phenotypic variation. Since the completion of the Human Genome Project, numerous studies of behavioral traits have directly measured DNA variation among individuals in an attempt to take this logical next step. While there are many types of genetic variants, most studies have assayed single-nucleotide polymorphisms (SNPs), which are sites in the genome where single DNA base pairs carried by distinct individuals may differ. 1 Virtually all SNPs have two different possible base pairs, called alleles. The less frequent allele in the population is called the minor allele of the SNP. If the frequency of a SNP’s minor allele in the population exceeds 1%, the SNP is called a common variant. Among individuals of European descent, there are approximately 8 million common variants in the human genome.
The results of studies searching for SNP-behavior associations have disappointed any hope that a small number of these common variants are responsible for a large percentage of cross-sectional trait variability. Instead, the evidence to date is consistent with what we propose as the Fourth Law of Behavior Genetics:
4. A typical human behavioral trait is associated with very many genetic variants, each of which accounts for a very small percentage of the behavioral variability. 2
For purposes of the law, a “typical human behavioral trait” is one that is (a) commonly measured by psychometric methods, (b) a serious psychiatric disease, or (c) a social outcome, such as educational attainment, that is plausibly related to a person’s behavioral dispositions. As is customary in behavioral science, we use the word law to describe what we consider to be a very robust empirical regularity (not a universal, mechanistic truth). In the remainder of this article, we will summarize the mounting empirical evidence in favor of the Fourth Law, consider what gene-behavior research strategies are likely to be profitable in light of the Fourth Law, and briefly consider possible explanations for the Fourth Law.
Empirical Evidence for the Fourth Law
Each person inherits two copies of any DNA segment, one from each parent, and therefore may carry 0, 1, or 2 copies of the minor allele at a particular SNP. The number of minor alleles can be taken to characterize the individual’s genotype at that SNP. The straight line that best fits the overall genotype-phenotype relationship is called the average effect of gene substitution (Fisher, 1941; J. J. Lee & Chow, 2013). The true genotype-phenotype relationship will certainly not be precisely linear, but the slope of the best-fitting straight line is equal to a weighted average of the phenotypic changes following from the possible gene substitutions. In principle, it is also possible to estimate the nonlinear effects of genotype, as well as gene-gene and gene-environment interactions. In practice, however, given the staggering combinatorial explosion of possible hypotheses, it will normally be a useful first step to estimate the average effects in order to identify a subset of SNPs that should be studied in greater detail (e.g., Rietveld, Esko, et al., 2014).
For example, a SNP designated “rs9320913” is located on chromosome 6 and has two alleles: C (cytosine) and A (adenine). It was identified in a recent genome-wide association study (GWAS) of educational attainment (Rietveld et al., 2013). A GWAS is a hypothesis-free analysis of the predictive power of each of approximately 1 million (or more) individual SNPs spread across the genome. The goal of a GWAS is to discover which SNPs are associated with a trait of interest (e.g., educational attainment, intelligence, extraversion, or schizophrenia). Because so many statistical tests are performed in a GWAS, the significance threshold is typically set at a stringent 5 × 10−8 (“p < .00000005”) rather than the 5 × 10−2 (“p < .05”) that is standard for behavioral studies assumed to be testing a single hypothesis; this practice is analogous to a Bonferroni correction. The association between rs9320913 and education reached the GWAS significance threshold and was also replicated at a conventional level in two follow-up studies of separate samples (Rietveld, Conley, et al., 2014; Rietveld et al., 2013).
Strikingly, each additional copy of A (the education-increasing allele) is associated with only one additional month of schooling. Note that a combined sample size of 126,599 participants from over 50 cohorts in 15 countries was used to discover and initially replicate this gene-education association; an additional sample of 34,428 participants was used for a second replication. The SNP rs9320913 is estimated to account for only 0.02% of the overall variability in educational attainment, but biometrical studies show that the total percentage of variability owed to genetic differences is three orders of magnitude larger (Heath et al., 1985; Rietveld et al., 2013). Since the SNPs with the largest effects are the easiest to find, these results suggest that educational attainment is a phenotype affected by thousands of undiscovered genetic variants, each responsible for a minuscule fraction of individual differences.
The story is similar for the better-studied phenotype of schizophrenia. Studies of DNA from over 36,000 diagnosed cases and 113,000 controls have so far identified 108 SNPs associated with schizophrenia at a strict evidentiary threshold, yet these 108 SNPs jointly account for only 3.4% of the variance of the trait (measured on a liability scale, with liability being an unmeasured but inferred trait that is normally distributed and must exceed a threshold in order for the measured trait—here, the diagnosis of schizophrenia—to occur; Ripke et al., 2014). Each of these 108 “hits” has a small effect size—typically less than a 1.1-fold increase in the odds of a schizophrenia diagnosis with each additional risk-conferring allele.
As GWAS sample sizes get larger, will more variants (presumably with even smaller effect sizes) be identified? The answer is surely yes, since the fraction of trait variance captured by currently known SNPs falls well below the traits’ heritabilities. Heritability—the total fraction of variance in a trait attributable to genetic factors—is traditionally estimated using behavioral-genetics methods. In recent years, however, a novel technique has been developed that uses GWAS data from nominally unrelated individuals to estimate an even more relevant benchmark: the total fraction of variance in a trait that is attributable to the average effects of SNPs (J. J. Lee & Chow, 2014; S. H. Lee, Wray, Goddard, & Visscher, 2011; Speed, Hemani, Johnson, & Balding, 2012; Yang et al., 2010). This technique is called genomic-relatedness-matrix restricted maximum likelihood (GREML). 3 In essence, GREML determines whether the randomly arising genetic similarity between pairs of unrelated individuals predicts the phenotypic similarity between those individuals; a stronger relationship between genetic and phenotypic similarity implies a greater influence of the measured SNPs on the trait of interest. Importantly, the magnitude of this influence can be accurately estimated even if the sample size is too small to identify all (or any) of the associated variants at a high level of confidence. GREML is perhaps the most important innovation in quantitative genetics to have been introduced in the last dozen years, and it has provided convincing evidence that the heritabilities of many psychological traits are more or less accurately estimated in traditional family studies (e.g., Chabris et al., 2012; Cross-Disorder Group of the Psychiatric Genomics Consortium, 2013; Davies et al., 2011; S. H. Lee et al., 2011; Rietveld et al., 2013).
It is also noteworthy that even if few individual SNPs contributing to the GREML-estimated heritability meet the evidentiary threshold of a hit, it is possible to use summary statistics from GWAS results to estimate coarse-grained quantities such as the total number of SNPs that will ultimately be found to be associated with the trait (Stahl et al., 2012). Applying this method to a subset of the schizophrenia data produced an estimate of 8,300 trait-associated SNPs (Ripke et al., 2013). Figure 1 demonstrates that the number of schizophrenia-associated SNPs discovered has increased substantially as GWAS sample sizes have increased. The results of Ripke et al. (2013) and the GREML studies cited above tell us is that there is every reason to expect the trend depicted in Figure 1 to continue.
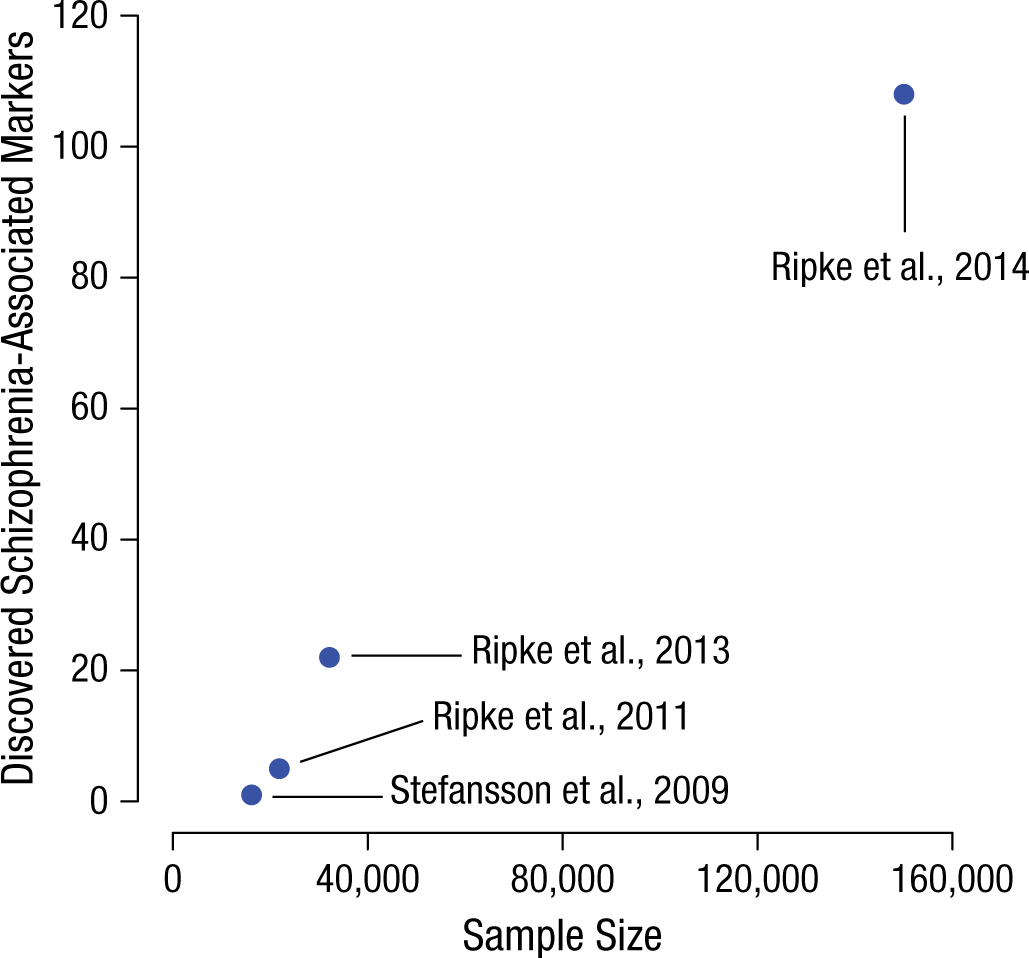
Number of schizophrenia-associated single-nucleotide polymorphisms clearing the strict genome-wide association study significance threshold (p < 5 × 10−8) as a function of discovery-stage sample size, which has increased over time. Although the four studies presented are not methodologically identical (because of different ratios of cases to controls), the increasing number of “hits” with sample size is nevertheless informative.
The genetic architectures of many human phenotypes tend to be highly polygenic (influenced by many different variants; Gottesman & Shields, 1967; Visscher, Brown, McCarthy, & Yang, 2012), and the Fourth Law states that this is also true for behavioral phenotypes. Since the number of common variants is finite (albeit large), the Fourth Law suggests in turn that the genetic architectures of human phenotypes are also pleiotropic (each variant affects many different traits). Moreover, evidence to date implies that behavioral phenotypes are even more polygenic than typical physical and medical traits. For example, the SNPs that have the largest effects on education (Rietveld et al., 2013) account for roughly one-tenth as much variability (0.02%) as those with the largest effects on physical traits, such as height and body mass index (roughly 0.3%). Moreover, as shown in Figure 2, a comparison of schizophrenia with a number of physical diseases suggests that its genetic variability is distributed among a greater number of variants with smaller effects (Ripke et al., 2013).
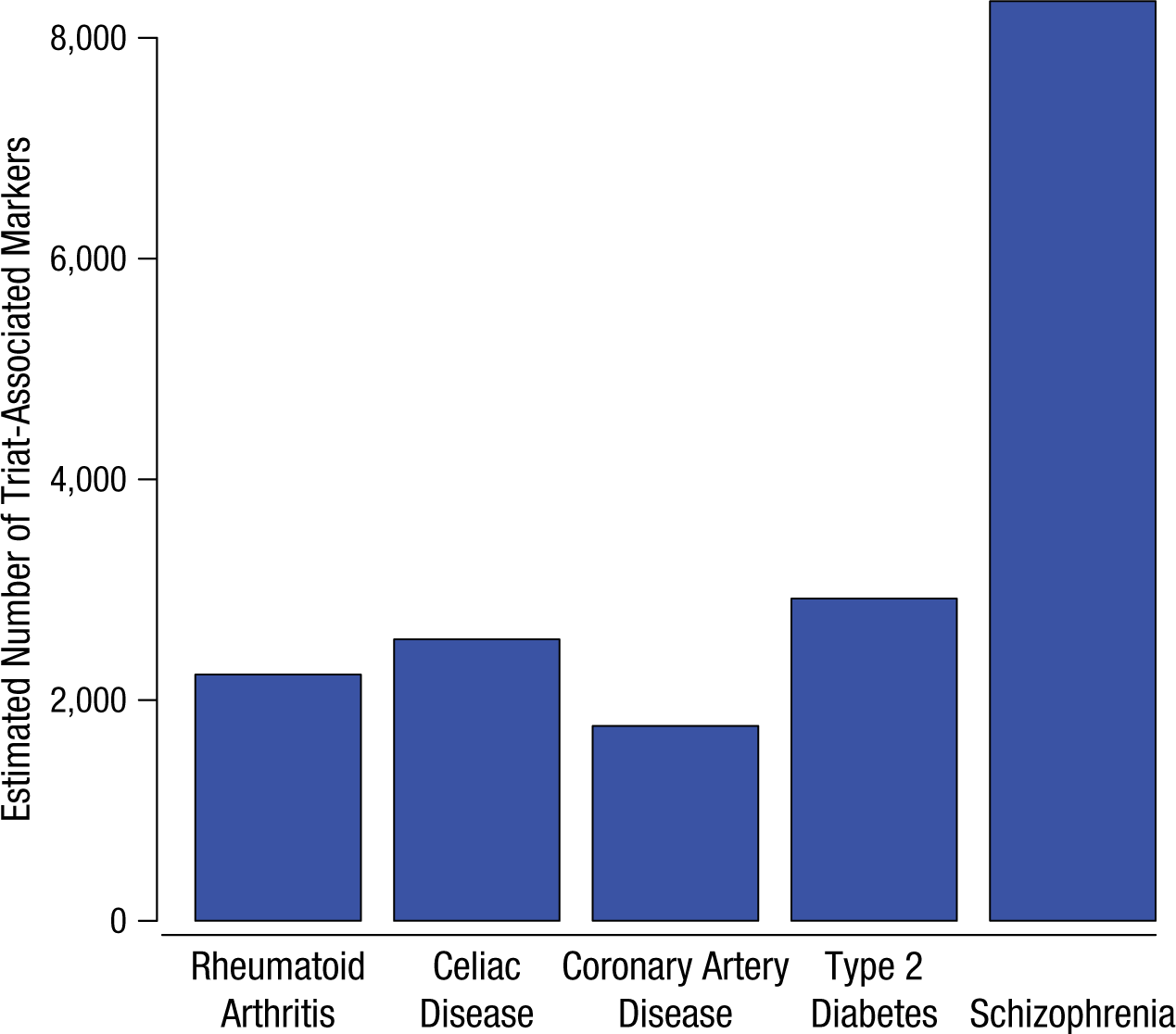
Estimated total number of single-nucleotide polymorphisms associated with five disease phenotypes based on results of genome-wide association studies (data drawn from Ripke et al., 2013; see that paper and Stahl et al., 2012, for further methodological details).
Besides the small effect sizes of positive results in well-powered (large-sample) studies, null results (at the 5 × 10−8 level) in less powerful studies provide converging evidence that variants with large effects are unlikely to exist. For example, null results have been obtained in GWAS of personality (de Moor et al., 2012), suggesting that these traits are not exceptions to the pattern described here.
It is possible that rare SNPs (with <1% frequencies of the minor allele) make an important contribution to the heritabilities of behavioral traits; such SNPs are not well represented in the GWAS we have cited. However, recent studies comprehensively assaying both common and rare variants in certain regions of the genome have failed to find any variants accounting for large portions of phenotypic variability (e.g., Purcell et al., 2014). Thus it seems that the inclusion of more variants in whole-genome sequencing studies will not alter the conclusion that individual genetic polymorphisms with effects on cognition, personality, education, or psychiatric disease accounting for even 1% of the variability are unlikely to exist. At this point, claims to the contrary should be considered extraordinary, and require corresponding amounts of evidence.
The Fourth Law also explains why the results of “candidate-gene” studies, which focus on a handful of genetic variants, usually fail to replicate in independent samples. The main problem is that such studies tend to have insufficient statistical power. If well-powered studies that search the entire genome for associations find only tiny effects, then large effects found in studies with sample sizes in the dozens to hundreds (e.g., Kogan et al., 2011; Skafidas et al., 2012) are likely to be false positives. This was shown empirically for the trait of general intelligence (g) by Chabris et al. (2012), who, using a sample of about 10,000 participants, failed to replicate published associations between g and 12 genetic variants. Accordingly, results of small-sample genetic studies should be regarded with great caution, especially in studies claiming to have identified interactions (Duncan & Keller, 2011).
However, candidate-gene studies can succeed when the sample is large and the candidate variants to be investigated have high prior probabilities of being associated with the trait—for example, when they consist of hits from a previous GWAS of a “proxy phenotype” that is itself strongly associated with the trait of interest. For example, Rietveld, Esko, et al. (2014) began with 69 SNPs that were associated with educational attainment (in a subset of the data from Rietveld et al., 2013) and tested them for association with g (which is correlated with educational attainment) in a separate sample of 24,189 individuals. Three of those SNPs were significant hits after adjustments for multiple hypothesis testing, representing the first robust discovery of common genetic variants associated with normal-range, non-age-related variation in general cognitive ability.
There are caveats to the use of the causality-implying term “effect” when discussing GWAS. One is that a SNP-phenotype association may reflect the causal effect of a correlated but unmeasured variant in the same genomic region. For example, if rs9320913 turns out to be a mere proxy for the causal variant, then the average effect of the true causal SNP on education may be somewhat larger than estimated by Rietveld et al. (2013). It is natural to wonder whether the empirical findings summarized by the Fourth Law might be artifacts attributable to the attenuation of larger effects caused by unmeasured variants, but careful investigation has shown that a multitude of causal variants with small effects must still be invoked to explain GWAS results (Wray, Purcell, & Visscher, 2011).
The Relevance of GWAS in Light of the Fourth Law
It has been argued that the empirical regularity summarized by the Fourth Law strengthens the case for deemphasizing gene-mapping studies (Turkheimer, 2012). We believe that the appropriate response to the Fourth Law is instead to pursue research strategies suited to the reality that most genetic effects on behavioral traits are very small.
Critics of GWAS findings have argued that they have a poor record of replication (McClellan & King, 2010; Turkheimer, 2012). It is true that the small SNP-trait association signals described by the Fourth Law are impossible to distinguish from noise in poorly powered studies. In our view, however, the Fourth Law makes clear that one solution is larger samples—much larger than most psychologists contemplate in the normal course of their research. Indeed, when GWAS are conducted with adequate sample sizes and strict evidentiary standards, the degree of quantitative replication is extremely strong (e.g., Rietveld, Conley, et al., 2014). 4 In a GWAS of height, the correlation among strictly significant hits between initial-sample and replication-sample estimates of SNP effects was nearly .97 (Lango Allen et al., 2010)—almost perfect agreement. Such precision holds even when comparing groups of different geographic origin (see Fig. 3). The correlation between effects measured in European and East Asian samples is only about .75 because the disease studies represented in Figure 3 employed smaller sample sizes than the height study. Nevertheless, it is evident that the best-fitting straight line in Figure 3 would approximate an intercept of 0 and a slope of 1, as would be expected if European effect sizes were estimated with no error and were equal to East Asian effect sizes (accounting for some sampling noise). This concordance is particularly remarkable because East Asians differ from Europeans in genetic background and environmental exposures. Evidence from studies of schizophrenia (de Candia et al., 2013; Ripke et al., 2014) suggests that this pattern will generalize to behavioral traits.
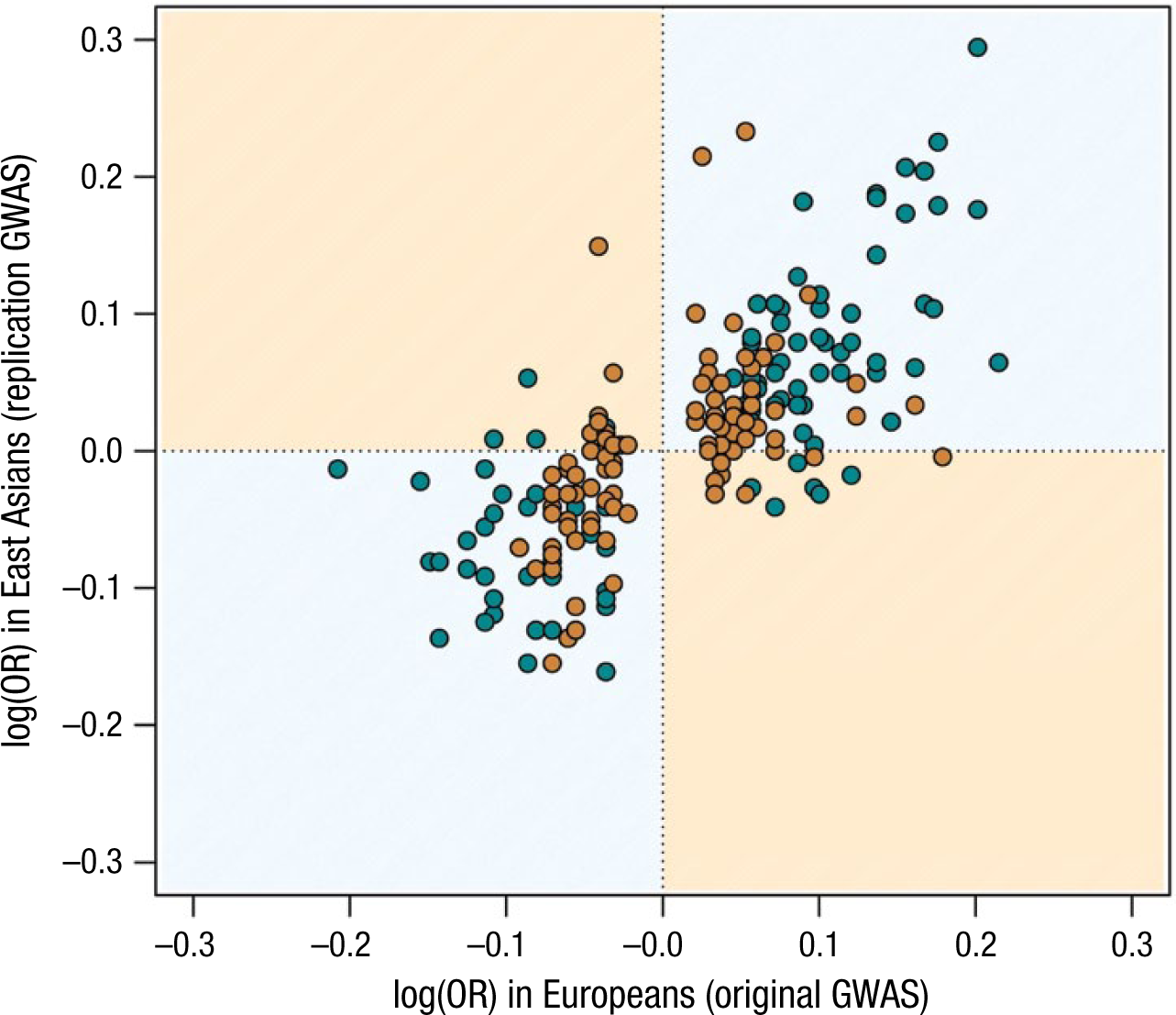
The strong agreement between genetic effects on 28 diseases estimated in Europeans and East Asians. The x-axis corresponds to the increment in the odds (on a logarithmic scale) of suffering from the disease for each additional copy of the reference allele, as estimated in Europeans. The y-axis corresponds to the same quantity estimated in East Asians. The increment in log(odds ratio [OR]) is the equivalent of the “additive effect” for dichotomously scored traits such as disease status (affected vs. unaffected). The gap in the data on the x-axis results from the fact that nonsignificant genetic effects would have odds ratios near 1 (and thus logarithms of 0) and therefore would not be included in the results of European samples. Green points are associations discovered in small genome-wide association studies (GWAS; less than 10,000 participants); orange points are associations discovered in large GWAS (more than 10,000 participants). Adapted from “High Trans-Ethnic Replicability of GWAS Results Implies Common Causal Variants,” by U. M. Marigorta, and A. Navarro, 2013, PLoS Genetics, 9, Article e1003566. Copyright 2013 by the authors. Adapted with permission.
Skeptics also caution that the problem of distinguishing causation from correlation in GWAS may be intractable (Charney, 2012; Turkheimer, 2012). Since observational studies can usually reveal only correlation, what is the justification for inferring that a SNP-phenotype association reflects an effect on the trait? Here we consider the issue of whether we can be confident that an association signal from some genomic region reflects the causal effect of a gene in that region rather than confounding with trait-affecting environmental conditions (J. J. Lee, 2012).
One major potential confound of this type is population stratification: If a SNP is more common in individuals of a certain ancestry or region, then it may falsely appear that the SNP is associated with a trait when the trait is actually associated with a pattern of ancestry or region of origin. Modern GWAS of unrelated individuals seek to account for this possibility by controlling for several principal components of the massive correlation matrix of all the assayed SNPs. These principal components typically identify the geographic ancestry of individuals in the sample (Price et al., 2006). There is a more elegant study design, however, that in follow-up studies can provide powerful evidence against confounding as an explanation for genetic associations.
When an organism becomes a parent, it passes on a randomly chosen allele from each of its pairs to a given offspring. Because the offspring’s genotype is randomly assigned conditional on the parental genotypes, a significant association between the presence of a particular allele and the focal phenotype within families is strong evidence for the presence of a nearby causal variant (Ewens, Li, & Spielman, 2008; Fisher, 1952; J. J. Lee & Chow, 2013). And indeed, family-based genetic studies have so far affirmed the results of GWAS that use unrelated individuals. For example, alleles identified as increasing liability to schizophrenia in standard GWAS were more likely, in a separate sample of families, to be transmitted from parents to affected children (Ripke et al., 2014; see also Benyamin, Visscher, & McRae, 2009; Lango Allen et al., 2010; Turchin et al., 2012; Wood et al., 2014). There is also evidence that the top SNPs identified by Rietveld et al. (2013) are jointly predictive of sibling differences in educational attainment (Rietveld, Conley, et al., 2014).
It has been argued that the effect sizes discovered by GWAS are so small that they are scientifically inert (e.g., Turkheimer, 2012). However, a small effect size from variation across individuals in a gene does not rule out a major qualitative role of the gene product itself in the relevant biological pathway. The principle is that making a small change to one step in a process may cause only a small change in the final output of the process, but making a large change, or omitting the step entirely, can halt the entire process. For example, by using molecular biological techniques to silence zebrafish genes whose orthologs were discovered in a GWAS to contain variants with small effects on human platelet count, researchers were able to abolish platelet production in this model organism (Gieger et al., 2011). Whether such cases will be numerous or rare for behavioral traits is a question that future GWAS-based research may soon answer.
Moreover, even though individual genetic variants have small effects, GWAS results can be used to construct a polygenic score, a variable that exploits the joint predictive power of many variants and therefore can explain substantial variance in the trait or outcome. The simplest polygenic score for a trait is constructed by adding up the individual effects measured for all of the SNPs in a GWAS (Dudbridge, 2013). Rietveld et al. (2013) found that such a polygenic score for educational attainment explains 2% to 3% of the variation across people—one hundred times more than was explained by the most predictive individual SNP. The power of polygenic scores increases with the size of the GWAS samples they are derived from; for instance, 15% of the variance in educational attainment could be explained if the scores came from a sample of 1 million individuals (Rietveld et al., 2013). Polygenic scores could have value for predicting disease or disability risk (Ripke et al., 2014), for identifying individuals who might benefit from early treatment or intervention, for studying gene-environment interactions, and for modeling genetic differences among individuals in epidemiological and experimental studies of behavioral and biological treatments (Rietveld, Conley, et al., 2014; Rietveld et al., 2013).
When the early GWAS of height were published, the confirmed SNP associations had a combined explanatory power of only 2% to 3%. This low predictive power was thought by some to illustrate fundamental limitations of GWAS methodology, such as its inability to estimate interactions and nonlinear effects that were hypothesized to account for the apparently “missing heritability.” This may turn out to be true for some phenotypes, but GREML studies have now confirmed that for many phenotypes, including height, intelligence, and schizophrenia, the combined additive effects of common SNPs do account for a large fraction of the variance. 5 The most recent GWAS of height (Wood et al., 2014) brought this fraction to about 17% (if the criterion is out-of-sample predictive accuracy) and 29% (if the criterion is GREML-estimated heritability attributable to the 10,000 SNPs with the strongest evidence for association). Thus, much of the heritability of height was not “missing” but merely hiding in the form of small but additive effect sizes. This can be seen as yet another illustration of the Fourth Law in action.
The Importance of the Fourth Law
We have recently highlighted two possible explanations for the Fourth Law (Chabris et al., 2013). First, causal chains from DNA variation to behavioral phenotypes are likely very long (longer than for physical traits—e.g., eye color), so the effect of any one variant on any one such trait is likely to be small. For example, a SNP may have a substantial effect on the concentration of an enzyme in cortical synapses, but that proximal biochemical phenotype is only one of many factors that explain why some people score higher than others on paper-and-pencil IQ tests, so the SNP has only a tiny effect on the distal phenotype of cognitive function.
Second, when a population is already well adapted to its environment, mutations with large effects on a focal trait are likely to have deleterious side effects (Fisher, 1930). If the effect of a genetic variant is small enough, however, then its population frequency has some chance of drifting upward to a detectable level (Kimura, 1983). 6 These forces could conspire to keep variants that have a large effect on a trait at negligible frequency while allowing some variants that have a small effect to become relatively common. Some support for this hypothesis comes from two observations: First, SNPs discovered in GWAS that have larger additive effects tend to have lower frequencies of the minor allele (Park et al., 2011), and second, variants with very large phenotypic effects, such as those causing mental retardation, are always very rare and thus contribute little to overall population variability, or they have their effects later in life (as in the case of, e.g, the well-documented relationship between variants of the apolipoprotein E, or APOE, gene and cognitive decline). These examples suggest that valuable evolutionary insights might flow from the detailed analysis of GWAS data (Turchin et al., 2012).
In conclusion, we shall place the Fourth Law in the context of what has long been well understood about the relationship between genes and human behavior—namely, that it is mistaken to believe that there might be a gene “for” one complex trait or another (for an eloquent statement of this basic point, see Dawkins, 1979, p. 189). What the Fourth Law adds to this understanding is that most genetic variability in behavior between individuals is attributable to genetic differences that are each responsible for very small behavioral differences.
The law we have proposed here provides a unified conceptual explanation for several consistent patterns in the results of the past two decades of gene-discovery studies, including the failure of candidate-gene studies to replicate, the need for GWAS (and why they actually do replicate), and the crucial importance of extremely large samples in these endeavors. We believe that compelling motives for pursuing gene-mapping studies of behavioral traits can be found in the promise of learning more about the evolutionary trajectory of the human species, the formulation of new biological hypotheses regarding cognition and neural function, and the value of polygenic scores in the social and medical sciences. The Fourth Law of Behavior Genetics provides fundamental guidance for how research in all of these areas can most efficiently progress.
Footnotes
Declaration of Conflicting Interests
The authors declared that they had no conflicts of interest with respect to their authorship or the publication of this article.
Funding
This work was supported by the Pershing Square Fund for Research on the Foundations of Human Behavior, Ragnar Söderberg Foundation Grant E9/11, Swedish Research Council Grant 412-2013-1061, and National Institute on Aging Grants P01AG005842, P01AG005842-20S2, P30AG012810, R01AG021650, and T32AG000186-23. The content of this publication is solely the responsibility of the authors and does not necessarily represent the views of any of these funding organizations.