
Abstract
Hematopoietic stem cell transplantation (HSCT) is frequently accompanied by severe inflammation-related complications, among which graft-versus-host disease (GVHD) stands out as one of the most common and life-threatening. As a systemic inflammatory disorder, GVHD arises when donor T cells recognize the recipient’s alloantigens and initiate an immune attack. Currently, effective second-line treatment options remain scarce for patients with antibiotic-resistant or steroid-refractory GVHD. Mesenchymal stem cells (MSCs) are non-hematopoietic cells widely distributed in fetal and adult tissues and organs, endowed with multipotent differentiation potential and prominent immunomodulatory properties. Emerging evidence suggests that impaired function or apoptosis of MSCs exacerbates HSCT-associated complications and significantly compromises hematopoietic stem cell engraftment. Over the past two decades, leveraging their potent anti-inflammatory and immunomodulatory capacities, MSCs have been rapidly integrated into HSCT research and clinical practice, where they play a pivotal role in promoting hematopoietic engraftment and preventing or treating GVHD. This review elaborates on the molecular basis of MSCs’ anti-inflammatory effects and the inflammatory pathological characteristics of HSCT-related complications. We conducted a systematic literature search in PubMed, Web of Science, and Embase databases up to December 2025. The search strategy combined the following terms: (“mesenchymal stem cells” OR “MSCs”) AND (“hematopoietic stem cell transplantation” OR “HSCT”) AND (“graft-versus-host disease” OR “GVHD”) AND (“inflammation” OR “immune regulation”). It systematically analyzes the molecular mechanisms underlying MSCs’ anti-inflammatory actions and their application progress in HSCT, aiming to provide a theoretical foundation and translational insights for the rational clinical application of MSCs in HSCT.
Keywords
Introduction
Hematopoietic stem cell transplantation (HSCT) is a fundamental therapeutic approach in the medical field for treating various hematologic malignancies, including acute leukemia, chronic myeloid leukemia, multiple myeloma, and lymphoma, as well as certain congenital or acquired immunodeficiency diseases, such as severe combined immunodeficiency and aplastic anemia with immune abnormalities. Since its introduction in clinical practice, HSCT has saved countless lives and significantly improved treatment outcomes for these conditions 1 . While HSCT offers curative potential for patients, it also comes with a range of severe complications that significantly affect their quality of life and can be a major factor limiting their long-term prognosis. Among these complications, acute and chronic graft-versus-host disease (GVHD), inflammatory storms, and delayed hematopoietic reconstitution are the most common and detrimental 2 . GVHD is one of the most prevalent complications following HSCT, with an incidence rate ranging from 30% to 50%. It is categorized into two types: acute GVHD (aGVHD) and chronic GVHD (cGVHD), based on when the symptoms appear. Acute GVHD typically manifests within the first 100 days post-transplantation, and in severe cases, it can lead to multiple organ failure, resulting in a mortality rate between 15% and 40%. Chronic GVHD usually develops more than 100 days after transplantation and can affect nearly all organ systems in the body, presenting with complex and varied clinical symptoms. Some patients may even exhibit autoimmune-like symptoms, which can significantly impair their daily functioning and potentially lead to long-term disability or death 3 .
In clinical practice, traditional immunosuppressive agents such as cyclosporine A, tacrolimus, and methotrexate are essential for preventing and treating GVHD 4 . While these medications can help control the onset and progression of GVHD by inhibiting the activation and proliferation of immune cells, they also carry significant side effects, most notably an increased risk of infections 5 . These infections can worsen patients’ conditions and may interact with GVHD, creating a vicious cycle that further deteriorates prognosis. Another serious complication after HSCT is a cytokine storm, which typically occurs in the early post-transplant period. This condition is often linked to tissue damage caused by conditioning regimens such as chemotherapy and radiotherapy, immune reactions during graft engraftment, and infections 6 . If not managed promptly, a cytokine storm can escalate rapidly to septic shock and multi-organ failure, posing a serious threat to patients’ lives. In addition, transplant-associated thrombotic microangiopathy (TA-TMA) and hepatic veno-occlusive disease (VOD) are also severe complications that jeopardize the safety of HSCT patients7,8. The occurrence and development of these complications are closely related to the inflammatory microenvironment after HSCT, highlighting the importance and urgency of improving this microenvironment and finding safe and effective strategies for preventing and treating complications. These issues have become key clinical challenges limiting the improvement of HSCT efficacy.
Against this backdrop, mesenchymal stem cells (MSCs), a type of non-hematopoietic stem cell with multi-directional differentiation potential and strong immune regulatory functions, have gradually become a research hotspot in the medical field 9 . MSCs are widely distributed in various organs and tissues of fetuses and adults, such as bone marrow, umbilical cord, placenta, and adipose tissue. They are relatively easy to obtain and have low immunogenicity (e.g. low expression of major histocompatibility complex class II molecules and co-stimulatory molecules on their surface, making them less likely to trigger host immune rejection), laying a solid foundation for their clinical application 10 . MSCs exert powerful immune regulatory functions through the secretion of anti-inflammatory cytokines and exosomes, mitochondrial transfer and metabolic reprogramming, and direct interaction with immune cells 11 . Based on these unique biological functions, MSCs have attracted extensive attention in the research of various inflammation-related diseases (such as rheumatoid arthritis, ulcerative colitis, and acute respiratory distress syndrome) and in the field of HSCT, providing new ideas and strategies for the prevention and treatment of post-HSCT complications. In recent years, scholars both domestically and internationally have conducted extensive basic research and clinical studies on the application of MSCs in HSCT. They have made significant progress in preventing and treating GVHD, alleviating cytokine storms, promoting hematopoietic reconstitution, and improving complications such as TA-TMA and VOD.
This article reviews the research advancements related to MSCs in HSCT, covering both basic and clinical aspects. It emphasizes the molecular mechanisms behind their anti-inflammatory effects and addresses the challenges encountered in clinical translation. By thoroughly organizing and analyzing the existing research findings, the article aims to provide valuable insights for further in-depth studies on MSCs in the HSCT field, support their clinical translation and application, and ultimately enhance the prognosis and efficacy of HSCT for patients. A graphical overview of topics related to the research field is presented in Fig. 1, and specific details are further elaborated throughout the article’s thematic sections.

Graphical overview of the anti-inflammatory effects of MSCs and their research progress in HSCT. Created by the authors using BioRender and Microsoft PowerPoint.
The anti-inflammatory mechanisms of MSCs
Biological characteristics of MSCs
MSCs play a crucial role in regenerative medicine and immunotherapy due to their biological origin and differentiation potential. During embryonic development, MSCs originate from the mesoderm, which gives them the ability to differentiate into various cell types and possess strong immune regulatory functions 12 . As a key germ layer in embryonic development, the mesoderm allows MSCs to develop into cells that form mesodermal tissues. This capability makes MSCs essential for maintaining the body’s immune balance, positioning them as one of the most promising types of adult stem cells in contemporary medical research 12 .
MSCs possess unique biological characteristics that enhance their proliferation and differentiation capabilities. They exhibit strong telomerase activity, which effectively slows down the shortening of telomeres. This property allows for long-term stable proliferation and enables large-scale expansion of cells in vitro, which is essential for clinical applications 13 . MSCs proliferate primarily through asymmetric cell division, a process that ensures the maintenance of the MSC population while also providing an adequate supply of cells for differentiation into various cell types. Under suitable in vivo or in vitro induction conditions, MSCs can differentiate into several mesodermal-derived cell types, including osteoblasts, chondrocytes, adipocytes, and myocytes. Furthermore, with the appropriate regulatory microenvironments, they can differentiate into non-mesodermal-derived cells, such as neural cells (including neuron-like cells and glial cells) as well as hepatocyte-like cells 14 . This extensive differentiation potential makes MSCs invaluable for tissue repair and regeneration. For instance, in bone injury repair, MSCs can differentiate into osteoblasts, secrete bone matrix, and enhance mineralization of bone tissue, thereby accelerating fracture healing. In cases of post-myocardial infarction, MSC-derived heart muscle-like cells can partially replace damaged myocardial cells, improving myocardial contractile function and reducing ventricular remodeling 15 . In research focused on treating neurological diseases, such as Parkinson’s disease and spinal cord injuries, MSC-derived neural cells can restore damaged neural cells and facilitate the reconstruction of neural synapses, offering promising avenues for recovery of neural function 16 . MSCs exert anti-inflammatory effects through multiple pathways, including regulating immune cell function, secreting anti-inflammatory factors, and inhibiting pro-inflammatory signaling pathways 17 . In vitro experiments have shown that MSCs can inhibit the functions of T cells and B cells. Vaillant et al 18 . pointed out that MSCs suppress the proliferation and cytokine release of CD8+ T cells through intercellular contact and the secretion of soluble factors. Furthermore, due to their unique immunological properties, the immunologically immature state of MSCs can reduce immune responses, improve graft acceptance in allogeneic HSCT, and lower the risk of GVHD 19 .
A key biological characteristic of MSCs is their ability to “home” to specific target areas, making them ideal as targeted therapeutic carriers. Homing ability refers to MSCs’ capacity to detect chemotactic signals—such as inflammatory factors, chemokines, and indicators of tissue damage—released by tissues or cells during pathological events like injury, inflammation, or tumors. MSCs can migrate through the bloodstream or interstitial spaces and penetrate the vascular endothelial barrier of lesions by binding to adhesion molecules on the surfaces of endothelial cells, like VCAM-1 and E-selectin. Once they reach the target tissues, they accumulate specifically in these areas. Regulated by the local microenvironment, MSCs perform biological functions such as anti-inflammatory effects and tissue repair 19 . This “active targeting” property allows MSCs to overcome the limitations of traditional drugs, which often suffer from systemic distribution and low local concentration. As a result, MSCs enable precise delivery to lesion sites, improving therapeutic efficiency and minimizing systemic side effects. Furthermore, this ability can be harnessed to deliver various genes. For instance, Han et al. 20 used MSCs as carriers for tumor-targeted suicide genes to treat lung metastases, while Kim et al. 21 created MSC-IFNβ gene complexes, both achieving promising therapeutic outcomes.
The anti-inflammatory mechanisms of MSCs
Secretion of anti-inflammatory cytokines
The anti-inflammatory and immune regulatory functions of MSCs rely significantly on their extensive “cytokine secretion network.” By precisely releasing a variety of anti-inflammatory cytokines and immune regulatory factors, MSCs can effectively target and regulate the inflammatory microenvironment, inhibit excessive immune responses, and play a crucial role in treating various inflammation-related diseases 22 . These key factors are not secreted randomly; instead, they form a synergistic “cytokine combination,” which includes Tumor Necrosis Factor-α Stimulated Gene 6 (TSG-6), Prostaglandin E2 (PGE2), interleukin-10 (IL-10), indoleamine 2,3-dioxygenase (IDO), and hepatocyte growth factor (HGF). Each of these factors participates in regulating inflammation through unique molecular mechanisms. Under inflammatory stimulation, they create a dynamically adjusted “anti-inflammatory cytokine spectrum” that ensures precise and efficient inhibition of inflammatory responses 23 .
As one of the key anti-inflammatory factors secreted by MSCs, TSG-6 functions as an “early anti-inflammatory sentinel” in situations of inflammatory injury. Initially discovered to be inducibly expressed by TNF-α, TSG-6 primarily acts by binding to hyaluronic acid (HA) in the inflammatory microenvironment, forming an HA-TSG-6 complex. This complex significantly inhibits the excessive activation of the complement system, reduces the release of complement components, and prevents tissue damage mediated by the complement pathway 24 . In acute inflammatory conditions, such as acute lung injury and traumatic inflammation, TSG-6 secreted by MSCs can quickly reach the site of injury and block the “cascade amplification effect” of inflammation through the mechanisms mentioned above, thereby creating favorable conditions for subsequent tissue repair 25 . PGE2 is another crucial anti-inflammatory factor secreted by MSCs, and its role in regulating immune cell functions has been well established. Particularly, it inhibits T-cell activation and proliferation. In research focused on preventing and treating GVHD, blocking the PGE2 secretion pathway of MSCs—such as by using cyclooxygenase-2 inhibitors—significantly weakens their inhibitory effect on GVHD, confirming the central role of PGE2 in the anti-inflammatory actions of MSCs 26 .
Importantly, the cytokine secretion by MSCs is not static but forms a “dynamic response” under inflammatory stimulation. When MSCs detect pro-inflammatory signals in the inflammatory environment, intracellular inflammatory signaling pathways (such as NF-κB and MAPK) are activated, regulating the transcription and secretion of downstream anti-inflammatory cytokines. This activation leads to a significant increase in the expression levels of factors like TSG-6 and PGE2, creating a “unique anti-inflammatory cytokine profile” tailored to specific diseases 27 . This response mechanism—“the stronger the inflammation, the more secretion”—allows MSCs to match the intensity of inflammation precisely while avoiding potential side effects from excessive cytokine secretion in non-inflammatory states 28 .
From the perspective of signaling pathway regulation, the anti-inflammatory factors secreted by MSCs directly target and inhibit core pro-inflammatory signaling pathways, effectively achieving a “blockade at the source.” For example, PGE2 can phosphorylate IκBα, the inhibitory protein of NF-κB, by activating the EP2/EP4-cAMP-PKA pathway. This phosphorylation keeps IκBα in a phosphorylated state, preventing NF-κB from being released from the cytoplasm and entering the nucleus to initiate the expression of pro-inflammatory genes 29 . More importantly, MSCs can establish a “positive feedback anti-inflammatory loop” with the inflammatory microenvironment. Pro-inflammatory cytokines, such as TNF-α and IL-6, first activate MSCs, prompting them to secrete more chemokines (like CXCL12 and CCL2) and immunosuppressive factors (including PGE2, IDO, and IL-10). The chemokines attract additional MSCs and endogenous anti-inflammatory cells, such as T regulatory (Treg) cells and M2 macrophages, to the site of inflammation, further enhancing the secretion of anti-inflammatory factors. On the other hand, immunomodulatory factors inhibit the activation of pro-inflammatory cells, reduce the release of pro-inflammatory cytokines, and amplify the anti-inflammatory effect by promoting M2 macrophage polarization30,31. This positive feedback loop (Fig. 2) enables the anti-inflammatory effects of MSCs to “self-reinforce,” ensuring that these effects remain efficient even in the presence of persistent inflammation until the inflammation subsides and the microenvironment returns to homeostasis.
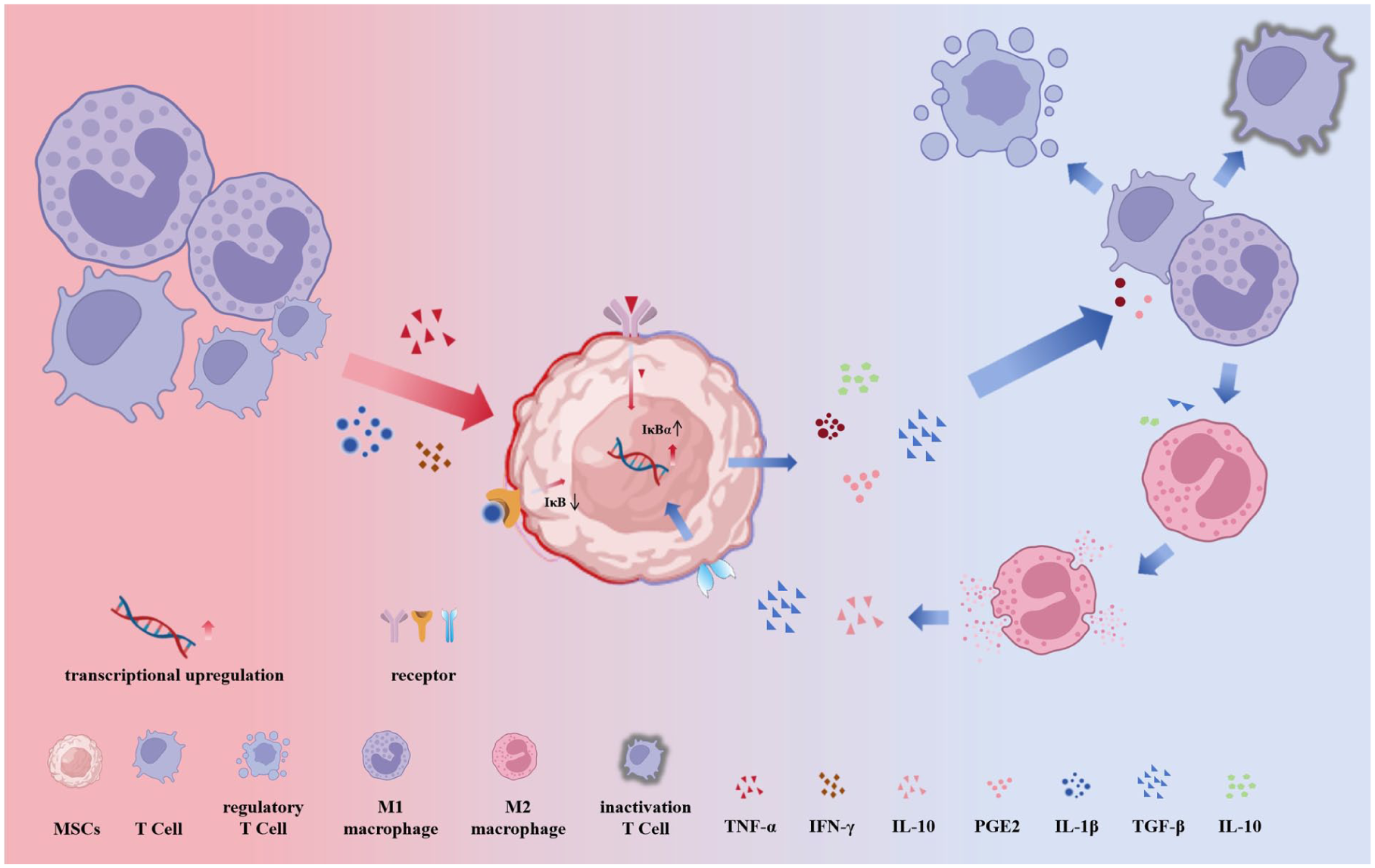
Dynamic anti-inflammatory cytokine secretion and positive feedback regulation of MSCs. In an inflammatory environment, activated T cells release TNF-α and IFN-γ, while macrophages secrete IL-1β and IL-6. These pro-inflammatory factors act on receptors (such as TNFR and IL-1R) on the cell membrane of MSCs, and reshape the immune microenvironment via the activated NF-κB signaling pathway (downregulating IκB, upregulating IκBα). (1) Inhibit inflammation: Reduce pro-inflammatory cytokines (TNF-α, IFN-γ, IL-1β) release; (2) Promote anti-inflammatory phenotype: Induce macrophage polarization toward M2 type, increasing anti-inflammatory factors (IL-10, TGF-β, PGE2); and (3) Regulate immune cells: Suppress T-cell activation and induce regulatory T-cell expansion, ultimately establishing an anti-inflammatory immune environment. Created by the authors using BioRender and Microsoft PowerPoint.
Mitochondrial transfer and metabolic reprogramming mechanisms
In recent years, researchers have identified mitochondrial transfer as an additional mechanism by which MSCs exert their anti-inflammatory effects. In an inflammatory microenvironment, MSCs can recognize damaged or overactive immune cells, such as CD4+ T cells, and transfer their functional mitochondria—capable of complete respiratory chain function and normal oxidative phosphorylation—into these target cells 32 . Under inflammatory conditions, CD4+ T cells often experience mitochondrial structural damage, including fragmented cristae and decreased membrane potential, or functional impairment characterized by insufficient ATP production and the excessive accumulation of reactive oxygen species (ROS). This damage typically results from oxidative stress and an increased metabolic load. However, the functional mitochondria derived from MSCs can quickly replenish the mitochondrial pool in the target cells, restoring their normal energy metabolism 33 . This process has two key benefits. First, it enhances ATP production to meet the energy demands needed for immune cell activation, thereby preventing dysfunction caused by energy deficiency. Second, it effectively inhibits the excessive activation of CD4+ T cells by clearing excess ROS and suppressing abnormal mitophagy activation. This action reduces the secretion of pro-inflammatory cytokines, such as IFN-γ and TNF-α, and hinders the escalation of inflammatory responses at their source34,35.
Mitochondrial transfer can occur through structures like tunneling nanotubes (TNTs) or extracellular vesicles (EVs). TNTs are tubular structures, measuring 50–1000 nm in diameter, that form through the extension of the cell membrane. They create direct cytoplasmic connections between MSCs and damaged cells, allowing for the active transport of mitochondria from MSCs to recipient cells. This process relies on the dynamic regulation of cytoskeletal proteins, such as microtubules and actin, as well as mitochondrial membrane transport proteins like Miro1, ensuring high targeting efficiency 36 . In contrast, EV-mediated mitochondrial transfer involves MSCs packaging mitochondria into vesicles, known as Mitochondria-Enriched EVs, which are then released into the extracellular microenvironment via paracrine signaling. These vesicles bind to specific receptors on the surface of recipient cells, such as integrins and glycoproteins, and deliver mitochondria through processes like endocytosis or membrane fusion. Compared to TNTs, EV-mediated mitochondrial transfer can reach target cells over greater distances. In addition, the vesicle structure helps protect the mitochondria from external environmental damage, thereby enhancing their stability and survival time in vivo. Together, these two pathways (Fig. 3) ensure efficient mitochondrial transfer in various inflammatory microenvironments 37 .
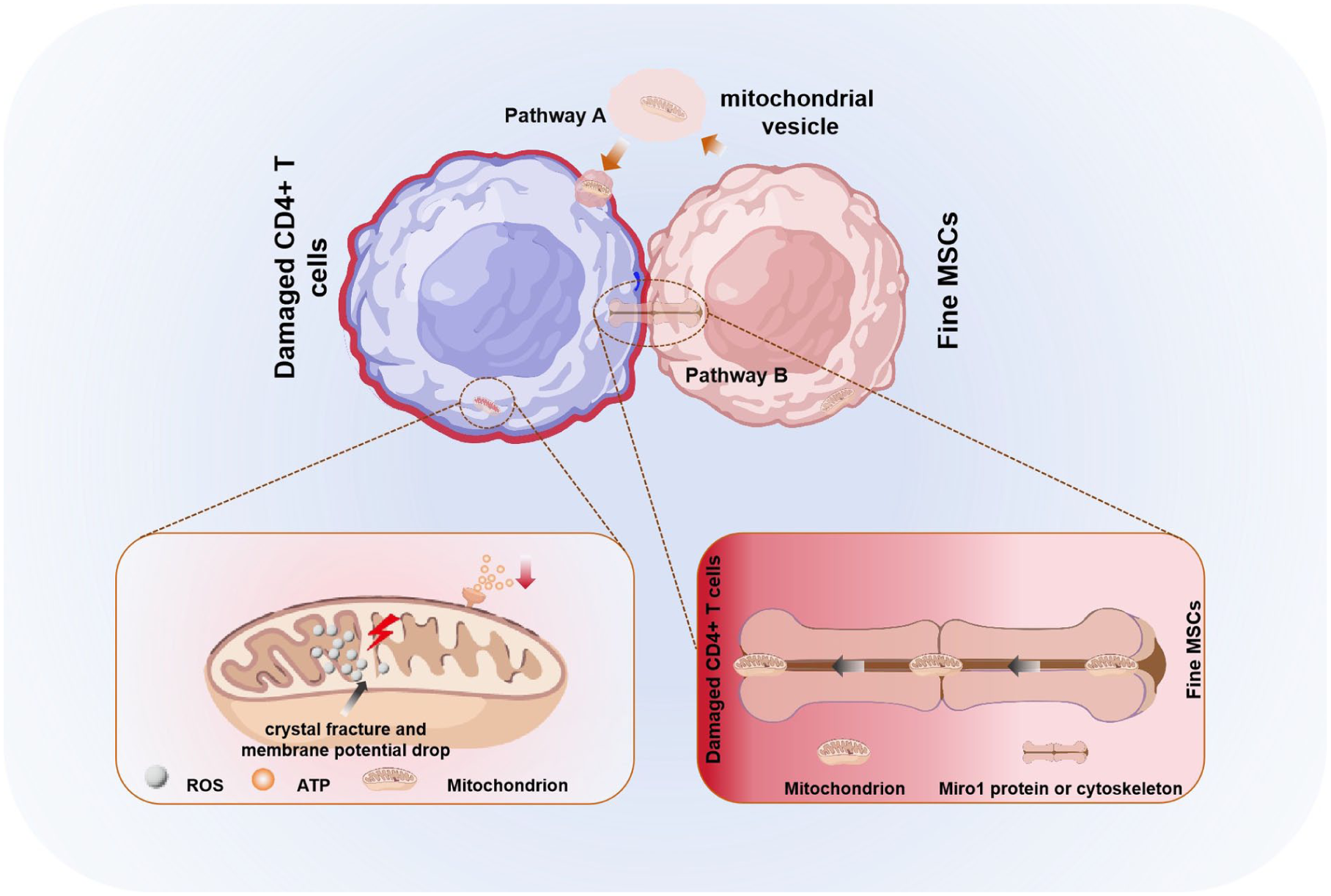
MSCs transfer functional mitochondria to metabolically impaired immune cells via tunneling nanotubes or extracellular vesicles, thereby restoring the latter’s functions. When CD4+ T cells are impaired, their internal mitochondrial structure is damaged, which manifests as the accumulation of ROS and insufficient ATP production. Pathway A: Via tunneling nanotubes, MSCs can transfer healthy mitochondria to impaired CD4+ T cells, a process driven by Miro1 protein and the cytoskeleton. Pathway B: MSCs release extracellular vesicles encapsulating healthy mitochondria. These vesicles fuse with the membrane of CD4+ T cells and deliver the mitochondria into the cytoplasm of impaired CD4+ T cells. Created by the authors using BioRender and Microsoft PowerPoint.
The therapeutic potential of mitochondrial transfer has been validated through various animal and cell experiments under specific pathological conditions. In models of acute pancreatitis, mitochondrial transfer by MSCs protects cells from damage caused by the abnormal opening of mitochondrial permeability transition pores (mPTPs) 38 . In models of pulmonary GVHD, damaged alveolar epithelial cells and pulmonary vascular endothelial cells release “damage signals” (such as HMGB1 and ATP) to recruit MSCs to migrate to lung tissues. Through mitochondrial transfer, MSCs provide protective effects in two key ways: First, they inhibit the excessive activation of donor CD4+ T cells, reducing the secretion of pro-inflammatory factors and alleviating T-cell-mediated immune damage. Second, they directly repair the mitochondrial function of alveolar epithelial cells and endothelial cells, decrease oxidative stress levels, and inhibit the process of epithelial-mesenchymal transition (EMT), which helps reduce lung tissue fibrosis 39 . Current studies have shown that pretreating MSCs to enhance their mitochondrial transfer capacity—such as by overexpressing the Miro1 protein—can further improve their preventive and therapeutic effects on pulmonary GVHD. This insight offers a novel strategy for treating pulmonary complications following HSCT 40 .
Exosome-mediated miRNA regulatory network
The anti-inflammatory effect of MSCs through the secretion of exosomes (MSC-Exo) is one of the important mechanisms of their immune regulatory functions. In terms of structural and compositional characteristics, MSC-Exo are single-layer membrane vesicles with a diameter ranging from 30 to 150 nm. Their membrane structure originates from the cell membrane of MSCs, thus retaining specific surface markers of the parent cells, such as the tetraspanin proteins CD9, CD63, and CD81. These markers are not only key criteria for identifying exosomes but also ensure the precise targeting of MSC-Exo to cells at inflammatory sites through specific binding with receptors on the surface of target cells 41 . Inside the vesicles, MSC-Exo encapsulate a variety of bioactive substances from MSCs, forming a “multi-functional regulatory system.” In addition to proteins (such as heat shock protein HSP70 and precursor proteins of anti-inflammatory cytokines) and lipids (such as ceramide and phosphatidylcholine involved in membrane signaling), nucleic acids such as microRNAs (miRNA) and nuclear RNAs (nRNA) are the core functional molecules exerting anti-inflammatory effects 42 . In addition to the targeted regulation by specific miRNAs, the reshaping of macrophage function by MSC-Exo is also an important supplementary mechanism for their anti-inflammatory effects. In treatments with exosomes from bone marrow-derived MSCs, upregulation of macrophage markers such as CD206 and anti-inflammatory cytokines can be observed, promoting the polarization of macrophages toward the M2 phenotype. At the same time, the production of ROS is reduced, inhibiting oxidative stress-induced cellular damage 43 . This regulation of macrophage polarization enables MSC-Exo to not only directly inhibit inflammatory responses but also accelerate the healing of damaged tissues by promoting the anti-inflammatory and reparative functions of macrophages, forming a synergistic “anti-inflammatory-repair” effect.
The specific miRNAs enriched in MSC-Exo target different inflammatory signaling pathways, forming the core mechanism network of their anti-inflammatory effects. Among them, three miRNAs play particularly crucial roles (Fig. 4):
miR-21-5p: By targeting and inhibiting the activation of programmed cell death protein 4 (PDCD4), miR-21-5p precisely blocks the NF-κB pathway, thereby suppressing the release of pro-inflammatory cytokines. In an acute lung injury model, miR-21-5p delivered by MSC-Exo can precisely target alveolar macrophages, reducing the damage caused by macrophages to alveolar epithelial cells, ultimately alleviating pulmonary edema, hemorrhage, and inflammatory infiltration in lung tissue, and improving lung function44,45.
miR-146a: This miRNA downregulates the expression of the TLR4/MyD88/NF-κB signaling axis in the inflammatory environment, inhibiting excessive immune activation. In an LPS-induced sepsis model, after the infusion of MSC-Exo rich in miR-146a, the protein levels of TLR4 and MyD88 in peripheral blood mononuclear cells and peritoneal macrophages of mice significantly decreased, the phosphorylation level of NF-κB p65 was reduced, and the secretion of pro-inflammatory cytokines was decreased. In addition, the recovery of body temperature and weight in mice was accelerated, and survival rates were significantly improved46,47.
miR-181a-5p: By targeting the STAT3 pathway, miR-181a-5p regulates the balance between Th17 and regulatory T cells, promoting the differentiation of anti-inflammatory regulatory T cells. In an autoimmune arthritis model, after the infusion of MSC-Exo carrying miR-181a-5p, the proportion of Th17 cells in the synovial tissue of mouse joints significantly decreased, while the proportion of Treg cells increased. The secretion of IL-17 was reduced, and the secretion of IL-10 increased, leading to a significant alleviation of joint swelling and cartilage damage 48 .

Specific miRNAs enriched in MSC-Exo target various inflammatory signaling pathways. MSCs release exosomes containing the membrane-associated proteins CD9, CD63, and CD81, with the exosomes packed with miRNA molecules. Pathway A: By targeting and inhibiting the activation of PDCD4, miR-21-5p precisely blocks the NF-κB pathway, thereby suppressing the release of pro-inflammatory cytokines. Pathway B: MiR-146a downregulates the expression of the TLR4/MyD88/NF-κB signaling axis and reduces the production of pro-inflammatory cytokines by targeting the mRNAs of TLR4 and MyD88 in monocytes within the inflammatory microenvironment. Pathway C: By targeting the STAT3 pathway, miR-181a-5p regulates the balance between Th17 and regulatory T cells, promoting the differentiation of anti-inflammatory regulatory T cells. Both Pathway A and Pathway B can enhance the anti-inflammatory response in sepsis mouse models, leading to their recovery. Created by the authors using BioRender and Microsoft PowerPoint.
Moreover, MSC-Exo can be widely distributed throughout the body via the blood and lymph circulatory systems. Their membrane structure effectively protects internal active substances from degradation by nucleases and proteases in body fluids, ensuring they remain biologically active until reaching target cells, thereby laying the foundation for their subsequent effects 49 .
Mechanisms of direct interaction with immune cells
In the immunomodulatory network of MSCs, intercellular contact serves as a direct and precise mode of action. It works in synergy with the paracrine effects mediated by secreted factors to jointly regulate the function and differentiation of immune cells, playing an indispensable role in maintaining immune homeostasis and inhibiting excessive inflammatory responses. MSCs can inhibit the proliferation of T cells (especially Th1 and Th17 subsets) and the release of pro-inflammatory factors (e.g. IFN-γ, TNF-α) through intercellular contact, while promoting the differentiation of Treg cells and enhancing immune tolerance 50 .
Th1 cells mainly mediate cellular immunity by secreting pro-inflammatory factors such as IFN-γ and TNF-α, and are involved in pathogen clearance and autoimmune inflammation. Th17 cells promote neutrophil infiltration and tissue inflammatory damage by secreting IL-17 and IL-22 51 . When MSCs come into contact with Th1/Th17 cells, key molecules on their cell membrane bind to receptors on the T-cell surface, directly blocking the activation signaling pathways of T cells:
By binding to CD40L on the T-cell surface, the activation of the CD40/CD40L pathway is inhibited, reducing the “second signal” required for T-cell activation, thereby suppressing the proliferation of Th1/Th17 cells 52 .
MSCs express ligands for the Notch receptor (such as Jagged1 and Delta-like 1) on their surface. When these ligands bind to the Notch receptor on the surface of Th1/Th17 cells, they can regulate the activity of the Notch signaling pathway, blocking the pro-inflammatory functions of Th1/Th17 cells 53 .
In vitro experiments have shown that when MSCs are co-cultured with activated Th1/Th17 cells, and Transwell chambers are used to block direct cell-to-cell contact as a control, the proliferation rate of Th1/Th17 cells in the direct contact group is reduced by 40%–60% compared to the control group, and the concentrations of IFN-γ, TNF-α, and IL-17 in the cell culture medium are significantly decreased 54 . When cell-to-cell contact is blocked, the inhibitory effect of MSCs on Th1/Th17 cells is significantly weakened, proving that direct cell-to-cell contact is a key mechanism by which MSCs inhibit pro-inflammatory T-cell subtypes.
Treg cells, as “negative regulators” of immune regulation, inhibit the activation and proliferation of effector T cells through the secretion of anti-inflammatory cytokines such as IL-10 and TGF-β, thereby maintaining immune tolerance. MSCs provide “signaling support” for the differentiation of Treg cells through cell-to-cell contact, primarily by activating the TGF-β/Smad signaling pathway and signal transduction mechanisms mediated by HLA-G molecules. In a systemic lupus erythematosus model, after the infusion of MSCs, the proportion of Treg cells in the spleen and lymph nodes of mice significantly increased, and the secretion of IL-10 was enhanced, while the proportion of Th1/Th17 cells and the levels of pro-inflammatory cytokines decreased. If antibodies were used to block HLA-G or mTGF-β on the surface of MSCs, their ability to promote the differentiation of Treg cells was significantly weakened, further proving the key role of cell-to-cell contact in the regulation of Treg cells by MSCs.
MSCs can induce macrophages to shift from the pro-inflammatory M1 phenotype to the anti-inflammatory M2 phenotype, thereby reducing the secretion of pro-inflammatory cytokines such as IL-1β and IL-6, while increasing the release of anti-inflammatory cytokines like IL-10 55 . MSCs achieve this macrophage polarization shift by activating the CD200/CD200R pathway and regulating the ICAM-1/LFA-1 pathway 56 . MSCs can actively sense changes in the inflammatory microenvironment and dynamically adjust their immune regulatory capabilities through cell-to-cell contact mechanisms 57 . They also work in concert with secreted factors to form a multi-layered immune regulatory network. This interaction is bidirectional: not only can MSCs regulate the functions of immune cells, but the inflammatory signals released by immune cells can also act back on MSCs, enhancing their immune regulatory abilities 15 . This “positive feedback” mechanism ensures that MSCs can function efficiently at sites of inflammation.
The inflammatory pathological characteristics of HSCT-associated complications
The inflammatory cascade of GVHD
GVHD is the most common and life-threatening inflammation-related complication after HSCT. The pathological process of GVHD involves a complex inflammatory cascade, triggered by mature donor T cells attacking host thymic epithelial cells, causing severe tissue damage. This mechanism plays a critical role in the transition from aGVHD to cGVHD 58 .
The core mechanism of aGVHD occurrence is that activated donor T cells trigger an “inflammatory cascade,” accompanied by the massive release of pro-inflammatory factors and infiltration of immune cells 59 . During the pre-transplant chemo-radiotherapy conditioning phase, tissue damage leads to the release of “initiating factors” such as TNF-α and IL-1β, which activate antigen-presenting cells. Antigen-presenting cells further activate donor T cells, inducing them to secrete cytokines such as IFN-γ, IL-2, and IL-17, forming a positive feedback loop to exacerbate inflammation 60 . Inflammation mainly affects target organs such as the skin, intestines, and liver: basal cells of the skin epidermis undergo apoptosis, and lymphocytes (CD4+/CD8+ T cells) infiltrate the dermis, leading to skin damage; intestinal mucosal epithelium undergoes necrosis, atrophy, and shedding, with a large number of inflammatory cells (T cells, macrophages) accumulating in the lamina propria, accompanied by intestinal barrier damage that further amplifies inflammation, leading to diarrhea and protein loss; inflammatory factors damage the bile ducts in the portal area of the liver, and a large number of lymphocytes infiltrate the bile ducts, resulting in cholestasis and elevated bilirubin, which eventually progresses to cholangitis61–63.
Chronic GVHD is the main cause of death in patients without relapse of the primary disease after HSCT. The core mechanism of its occurrence lies in the coexistence of inflammation and post-inflammatory fibrosis, combined with the superposition of B cell activation, autoantibody production, and other factors, which disrupts tissue repair. In the early stage of cGVHD, lymphocyte infiltration in target organs is the primary feature; in the late stage, it is accompanied by the activation of fibroblasts, which secrete collagen fibers and lead to tissue fibrosis. During this process, B cells become overactivated, produce autoantibodies, and attack self-tissues; Th2 cells secrete cytokines such as IL-4 and IL-13, which promote eosinophil infiltration and fibrosis 64 . Unlike aGVHD, cGVHD mostly affects the oral mucosa (presenting with dryness and ulcers), lacrimal/salivary glands (manifesting as Sjögren’s syndrome-like features), and lungs (such as bronchiolitis obliterans) 65 . The degree of inflammation varies significantly depending on the organ and individual, exhibiting heterogeneity characterized by multi-organ involvement (Fig. 5).

The pathological cascade of multiple organ damage in post-transplant patients. After transplantation, immune cells (e.g. macrophages, lymphocytes) interact, triggering the initial inflammatory response. The inflammatory reaction induces the secretion of regulatory cytokines (IL-10, TGF-β). Antigen-presenting cells activate donor-derived T cells, amplifying the immune response. Activated cells release a massive array of cytokines, which is promoted by earlier regulatory factors. The cytokine storm drives widespread immune attack, causing damage to organs (e.g. liver, intestine, skin). Created by the authors using BioRender and Microsoft PowerPoint.
TA-TMA
TA-TMA is a rare yet devastating complication of HSCT, characterized by an incidence of 5% to 15% among recipients, with a mortality rate exceeding 60% in severe cases—making it one of the most life-threatening non-relapse complications post-transplant 66 . Rooted in profound vascular endothelial injury, TA-TMA’s core pathological hallmark lies in “vascular endothelial inflammatory damage,” a cascade that intricately intersects with dysregulated coagulation and complement activation, ultimately leading to microvascular thrombosis, end-organ ischemia, and progressive dysfunction. Unlike other HSCT-related complications, TA-TMA exhibits a distinct predilection for damaging the microvasculature of vital organs, with the kidneys being the most frequently and severely affected, though the brain, lungs, and gastrointestinal tract can also be involved 67 .
The pathogenesis of TA-TMA is multi-factorial, with the pre-transplant conditioning phase serving as a critical initiating trigger. High-dose chemotherapy (e.g. busulfan, cyclophosphamide) and total body irradiation—cornerstones of HSCT conditioning—directly induce oxidative stress and DNA damage in vascular endothelial cells, disrupting their structural integrity and physiological function 68 . Beyond direct cytotoxicity, secondary insults further amplify endothelial injury: acute graft-versus-host disease (aGVHD) triggers systemic inflammation that targets vascular endothelium, while post-transplant infections (viral, bacterial, or fungal) release pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) that activate endothelial cells via toll-like receptor (TLR) signaling. Once activated, endothelial cells undergo a phenotypic shift: they upregulate the expression of adhesion molecules (e.g. VCAM-1, E-selectin, ICAM-1) that recruit circulating leukocytes (neutrophils, monocytes) to the vessel wall and secrete procoagulant factors (e.g. tissue factor, von Willebrand factor [vWF]) while downregulating anticoagulant proteins (e.g. thrombomodulin, protein C) 69 . This pro-inflammatory, pro-thrombotic phenotype disrupts the delicate balance of the vascular microenvironment, setting the stage for subsequent pathological events.
The kidneys are the primary target organ of TA-TMA, reflecting their dense microvascular network and high susceptibility to microthrombotic injury 70 . Pathologically, renal TA-TMA is characterized by glomerular capillary microthrombi composed of platelets and fibrin, endothelial cell swelling and detachment from the basement membrane, and widening of the subendothelial space. These changes lead to impaired glomerular filtration, resulting in clinical manifestations such as proteinuria (often in the nephrotic range), hematuria, hypertension, and progressive acute kidney injury (AKI) 71 . If left untreated, the ongoing microvascular damage and ischemia can progress to irreversible renal fibrosis, culminating in end-stage renal disease (ESRD) that requires long-term dialysis or renal transplantation 72 . Notably, extrarenal manifestations may also occur, including neurological symptoms (e.g. headache, seizures, altered mental status) due to cerebral microthrombosis, gastrointestinal bleeding or ischemia, and pulmonary hypertension—all of which contribute to the high mortality of severe TA-TMA 73 .
Beyond the core cascade of endothelial injury, thrombosis, and inflammation, TA-TMA pathogenesis is further complicated by dysregulated complement activation and impaired MSCs function. A subset of TA-TMA patients harbors congenital or acquired deficiencies in complement regulatory proteins (e.g. CD55, CD59, factor H), which normally inhibit uncontrolled activation of the alternative complement pathway 67 . In these individuals, endothelial injury triggers excessive alternative complement pathway activation, leading to the formation of membrane attack complexes (MACs) that directly lyse endothelial cells and amplify vascular inflammation 74 . This complement-mediated damage not only exacerbates the core TA-TMA pathology but also impairs the survival and function of infused MSCs—cells with potent immunomodulatory and endothelial repair capabilities 75 . Preclinical and clinical evidence suggests that the pro-inflammatory, complement-activated microenvironment of TA-TMA reduces MSCs viability, inhibits their homing to damaged vasculature, and blunts their ability to secrete anti-inflammatory cytokines (e.g. PGE2, IL-10) and pro-angiogenic factors 27 . This impaired MSCs function creates a “therapeutic gap,” as the body’s intrinsic repair mechanisms are unable to counteract the ongoing endothelial damage, further propagating the disease course (Table 1).
Key pathological events in TA-TMA and corresponding MSCs intervention targets.

The inflammatory microenvironment of VOD
VOD is a liver-specific complication that occurs after HSCT, characterized by occlusive inflammation and fibrosis caused by damage to the endothelial cells of the hepatic small veins and sinusoids. If not intervened in a timely manner, it can progress to liver failure or even death. The incidence of VOD after HSCT is approximately 5%–20%, while the mortality rate among patients with severe disease exceeds 80% 76 . High-dose chemotherapy or total body irradiation prior to HSCT is the primary contributing factor, as these treatments can directly damage the sinusoidal endothelial cells and small venous endothelial cells of the liver, occluding the lumens of hepatic sinusoids and small veins 77 . After endothelial cell injury within the hepatic lobules in the periportal region, pro-inflammatory factors and procoagulant factors are released; neutrophils and macrophages infiltrate the periportal region, further damaging endothelial cells and hepatocytes, and forming a vicious cycle of “injury—inflammation—thrombosis.” Clinical symptoms such as jaundice, hypoalbuminemia, and ascites occur 78 . In the inflammatory microenvironment of VOD, cholangiocyte stem cells, as a liver-specific stem cell population, have significantly impaired regenerative capacity. Clinical data show that patients with VOD may experience a recurrence of inflammatory reactions when immunosuppressants are reduced, manifesting as recurrent fever accompanied by elevated inflammatory markers. Notably, MSCs in the hepatic microenvironment of patients with VOD exhibit a unique chemokine receptor expression profile, which may affect the therapeutic efficacy of exogenous MSCs 79 .
Research on the application of MSCs in HSCT
Promoting hematopoietic reconstitution
Delayed hematopoietic reconstitution is one of the complications of HSCT, which may lead to an increased risk of infection and bleeding, seriously affecting the quality of patients’ prognosis. MSCs can differentiate into structural components that form the hematopoietic niche, such as osteoblasts, adipocytes, and vascular endothelial cells. For example, osteoblasts are located on the surface of the endosteum, forming an “endosteal niche” that provides a quiescent environment for HSCs; vascular endothelial cells form a “vascular niche” that supports the proliferation and homing of HSCs80,81. Co-transplantation of MSCs and HSCs can reconstitute the bone marrow microenvironment. As an important component of bone marrow stromal cells, MSCs can repair the bone marrow niche damaged by chemotherapy or radiotherapy and provide a scaffold for the colonization and survival of HSCs, which can shorten the engraftment time of neutrophils and platelets by approximately 3–5 days. A systematic review of 47 clinical studies (2000–2025, n = 1777) showed that co-infusion of MSCs and HSCs can shorten the engraftment time of neutrophils and platelets to 13.96 days and 21.61 days, respectively, without increasing the risk of recurrence or GVHD 82 .
Mitochondrial or metabolite transfer mediated by Connexin-43 gap junctions can reduce the level of ROS in HSCs and accelerate bone marrow niche revascularization. Adhesive junctions formed by N-cadherin between the surface of MSCs and the surface of long-term HSCs maintain HSC quiescence and inhibit ROS-induced stem cell exhaustion 83 . Recent work shows that MSC-derived microvesicles upregulate Jagged-1 and Notch-2 expression in hematopoietic stem/progenitor cells, activating the Jagged-1-Notch axis and enhancing self-renewal while restraining differentiation 84 .
MSCs can secrete hematopoietic growth factors (such as granulocyte colony-stimulating factor, erythropoietin, and chemokines) to support the proliferation and differentiation of HSCs 85 . Stem cell factor, Fms-like tyrosine kinase 3 ligand, thrombopoietin, and other factors secreted by MSCs can directly stimulate the proliferation of HSCs and inhibit their apoptosis 86 . For example, researchers have demonstrated through clinical and experimental studies that co-transplantation of MSCs can increase the number of CD34+ cells in peripheral blood and confirmed that the binding of stem cell factor (SCF) to the c-Kit receptor on the surface of HSCs can maintain the survival and self-renewal of HSCs 87 . MSCs regulate the tendency of hematopoietic differentiation by secreting granulocyte colony-stimulating factor, erythropoietin, macrophage colony-stimulating factor, and so on, and directionally promote the differentiation and maturation of hematopoietic cells such as granulocytic lineage, macrophage lineage, and erythrocytic lineage. MSCs can also secrete insulin-like growth factor, transforming growth factor-β, and other factors to regulate processes such as glucose uptake and energy metabolism of HSCs, thereby affecting the metabolic state of the bone marrow microenvironment, providing appropriate nutritional support for HSCs, and promoting their growth 88 .
MSCs can promote the migration of HSCs from peripheral blood to the bone marrow hematopoietic niche and their colonization, which in turn facilitates the homing of HSCs. MSCs can promote the migration of HSCs from peripheral blood to the bone marrow hematopoietic niche and their colonization, which in turn facilitates the homing of HSCs. MSCs are capable of secreting chemokines: after HSCT, MSCs release stromal cell-derived factor-1 (SDF-1). MSCs are capable of secreting chemokines: after HSCT, MSCs release SDF-1, which binds to the CXCR4 receptor on the surface of HSCs to form a chemical gradient, guiding the migration of HSCs to the bone marrow and accelerating their colonization 89 . In 2013, animal experiments conducted by researchers confirmed that overexpression of stromal cell-derived factor-1 (SDF-1) in MSCs can accelerate platelet recovery 90 . Immunosuppression after transplantation tends to cause infections by bacteria, viruses, and other pathogens, triggering an inflammatory response in the body. Under the stimulation of inflammatory or injury signals, MSCs can upregulate the expression of adhesion molecules such as intercellular adhesion molecule 1 (ICAM-1) and vascular cell adhesion molecule 1 (VCAM-1), thereby enhancing the ability to capture and anchor homing HSCs 91 . Regulating bone marrow vascular permeability is one of the essential steps in the process of hematopoietic reconstitution. By secreting factors such as vascular endothelial growth factor (VEGF), MSCs can promote an increase in vascular permeability, extracellular matrix denaturation, migration and proliferation of vascular endothelial cells, and angiogenesis 27 . These effects lead to a transient increase in the permeability of bone marrow vascular endothelium, facilitating HSCs to cross the vascular wall and enter the hematopoietic niche (Table 2, Fig. 6).
Key mechanisms of MSCs in promoting hematopoietic reconstitution post-HSCT.


MSCs in the bone marrow regulate the hematopoietic microenvironment. The first part presents that MSCs can differentiate into vascular endothelial cells, Gegenbaur cells, which contribute to the formation of the bone marrow microenvironment. In this microenvironment, hematopoietic stem cells and osteoblasts are connected by N-cadherin. The second part presents that MSCs secrete various factors to regulate the microenvironment. The last part presents that MSCs bind to the Notch-1/2 protein receptors on HSCs via the Jagged-1/2 signaling pathway, thereby promoting HSC self-renewal. And MSCs connect with HSCs via gap junctions formed by Connexin-43, facilitating mitochondrial and metabolite transfer to reduce ROS levels within HSCs. Created by the authors using BioRender and Microsoft PowerPoint.
Prevention and treatment of GVHD
GVHD is one of the most severe complications after HSCT, triggered by donor T cells attacking the recipient’s tissues, and manifests as damage to multiple organs such as the skin, intestines, and liver. However, MSCs have shown significant potential in the treatment of GVHD following HSCT. The main mechanisms are as follows: MSCs can alleviate the inflammatory response of GVHD by inhibiting the activation of donor T cells, promoting the proliferation of regulatory T cells, regulating the cytokine balance (e.g. reducing TNF-α and increasing IL-10), and decreasing the release of perforin/granzyme by tissue-resident CD8+ T cells 92 .
Clinical studies have confirmed that bone marrow–derived MSCs exhibit significant efficacy in patients with aGVHD. Among relevant products, Ryoncil™ (remestemcel-L), a human bone marrow–derived MSC product, has entered the clinical evaluation phase 93 . As of 2020, more than 70 clinical trials of MSCs for steroid-refractory aGVHD have been completed or are ongoing worldwide 94 . Among these, the phase III results of Ryoncil™ (developed by Mesoblast) in 2024 showed that bone marrow–derived mesenchymal stem cells achieved an overall complete response (CR) rate of 30% and increased 6-month overall survival by 57% compared with historical controls 95 . Umbilical cord-derived MSCs have demonstrated enhanced therapeutic efficacy after interferon-gamma (IFN-γ) gene transfection. A study involving 86 patients with grade III–IV steroid-refractory aGVHD showed that umbilical cord mesenchymal stem cells treatment yielded encouraging clinical responses 96 . Prophylactic use of MSCs after haploidentical HSCT can significantly reduce the incidence of cGVHD. Studies have indicated that repeated infusions of umbilical cord mesenchymal stem cells in the early post-haploidentical HSCT period (on days +45 and +100) can effectively prevent the development of cGVHD 3 . This prophylactic strategy has also shown promising prospects in patients with severe aplastic anemia (Table 3).
Summary of key clinical trials on MSCs in HSCT.

To enhance the therapeutic efficacy of MSCs, researchers are actively exploring combined treatment regimens. The regimen of MSCs combined with basiliximab and calcineurin inhibitors, used as second-line therapy for steroid-refractory aGVHD, has demonstrated superior efficacy compared to monotherapy in randomized phase III clinical trials 97 . In the context of haploidentical transplantation, the regimen of MSCs combined with basiliximab for the treatment of steroid-refractory aGVHD has also achieved significant effects 98 . In addition, the combined use of MSCs and immunosuppressants can produce a synergistic effect, jointly inhibiting excessive immune responses through different mechanisms. Notably, exosomes derived from MSCs can mimic the biological functions of their parent cells, alleviating GVHD symptoms while preserving the anti-leukemic effect, providing a new perspective for the development of cell-free therapeutic strategies 99 (Fig. 7).

Clinical application of MSCs in the treatment of GVHD. The first part presents that early post-transplantation administration of MSCs derived from the placenta or bone marrow can reduce the incidence of cGVHD. The next part presents that aGVHD occurs in the early period after transplantation and the infusion of MSCs can effectively treat refractory aGVHD. The last part presents that the combination of MSCs with basiliximab plus calcium-dependent phosphatase inhibitors can effectively treat refractory aGVHD. Created by the authors using BioRender and Microsoft PowerPoint.
MSCs reduce the risk of infection after HSCT
After HSCT, patients’ immune defense functions are suppressed, making them prone to bacterial, viral, or fungal infections. The anti-inflammatory and immunomodulatory effects of MSCs can reduce the consumption of immune cells caused by excessive inflammation, while enhancing anti-infective capacity by promoting the functions of macrophages and natural killer cells 100 . MSCs can reduce the risk of post-transplant bacterial/viral infections by upregulating the antimicrobial peptide LL-37 and enhancing the phagocytic function of macrophages. In a 2025 clinical study on patients with severe COVID-19, results showed that MSCs significantly reduced the levels of C-reactive protein, TNF-α, and IL-6, and improved the oxygenation index—providing a reference for the combined treatment of post-transplant infectious complications 101 . In addition, antimicrobial peptides and other substances secreted by MSCs can also directly inhibit pathogens. In vitro experiments by Krasnodembskaya et al. demonstrated that human bone marrow–derived MSCs and their conditioned medium inhibited the growth of Escherichia coli by 40% and Pseudomonas aeruginosa by 70% within 6 h. This effect was dependent on the antimicrobial peptide LL-37 secreted by MSCs; when LL-37 was silenced with siRNA, the antimicrobial activity almost disappeared 102 .
MSCs secrete factors such as VEGF and fibroblast growth factor (FGF), which accelerate the healing of respiratory and gastrointestinal epithelium damaged by radiotherapy and/or chemotherapy, thereby reducing the chance of bacterial infection 103 . Latest clinical studies have found that MSC treatment can restore intestinal alpha diversity, reduce potential pathogenic bacteria, and indirectly lower the risk of bloodstream infection. Two HSCT-related prospective cohorts (n = 128 and n = 94) showed that prophylactic infusion of MSCs (1 × 106 cells/kg on days +1 and +8) reduced the incidence of bacteremia within 100 days after transplantation from 27% to 11%, and the incidence of fungal infection from 9% to 2%, with no MSC-related adverse reactions observed 3 . Without compromising the body’s immunity, MSCs significantly reduce the risk of infection after HSCT and may serve as a supplement or alternative to standard infection prevention protocols in the future.
Limitations and challenges of MSCs therapy in HSCT
Despite the promising preclinical and clinical evidence, several limitations hinder the widespread application of MSCs in HSCT. First, MSCs products exhibit significant heterogeneity due to differences in tissue origin (bone marrow, umbilical cord, adipose tissue), donor variability, isolation methods, and culture conditions. This variability leads to inconsistent therapeutic outcomes and complicates cross-study comparisons 104 . At present, the lack of unified cell culture standards and quality control systems results in significant differences in MSC products obtained from different research centers. In addition, cell senescence that may occur during conventional expansion and culture further exacerbates product heterogeneity 105 . Second, MSCs possess dual immunomodulatory effects—while they typically suppress inflammation, under certain pathological conditions (e.g. high concentrations of pro-inflammatory cytokines) they may paradoxically promote inflammation or contribute to fibrosis via the secretion of pro-fibrotic factors such as TGF-β 106 . This functional plasticity makes the prediction of clinical efficacy more complex. Third, the short in vivo survival time of infused MSCs (often <72 h) limits their sustained therapeutic efficacy. Most intravenously administered MSCs become trapped in the lungs and are cleared rapidly, reducing their ability to home to target tissues 107 . Fourth, the lack of standardized protocols for dosing, timing, and administration routes leads to heterogeneity in clinical outcomes. Optimal treatment regimens (e.g. single vs. repeated infusions, cell dose) have not been established 108 .
Fifth, many clinical studies suffer from small sample sizes, lack of control groups, and short follow-up periods, limiting the generalizability and long-term safety assessment of MSCs therapy. Finally, the functional plasticity of MSCs in response to the host microenvironment—especially in the context of inflammation, aging, or comorbidities—adds another layer of complexity. Future research should focus on identifying predictive biomarkers, optimizing cell manufacturing, and conducting well-designed multicenter randomized controlled trials to address these challenges.
Conclusion and additional future directions
MSCs have emerged as a highly promising adjuvant therapy in the field of HSCT due to their significant anti-inflammatory and immunomodulatory properties. Basic research has clarified that MSCs exert anti-inflammatory effects through multiple mechanisms, including the secretion of cytokines such as prostaglandin E2 (PGE2) and interleukin-10 (IL-10), the regulation of networks via miRNA transfer through exosomes, and direct interactions with immune cells. These findings provide a solid theoretical basis for clinical applications. Through the aforementioned mechanisms, MSCs demonstrate multiple potentials in HSCT, such as preventing GVHD, promoting hematopoietic reconstitution, and reducing the risk of infection. With the deepening of basic research and clinical trials, MSCs are expected to become an important tool for adjuvant therapy in HSCT, further improving the success rate of transplantation and the quality of life of patients.
In the future, an effective translational medicine research platform can be established to address the aforementioned limitations and deficiencies. For instance, organoid models and 3D culture systems can be developed to better simulate the in vivo microenvironment, and multi-omics technologies can be used to identify biomarkers that predict therapeutic responses. Focus can also be placed on genetic modification engineering: overexpressing specific cytokines can effectively promote the transformation of macrophages to an anti-inflammatory phenotype, thereby avoiding the dual anti-inflammatory and pro-inflammatory effects of MSCs. Recent studies have confirmed that the combination of the CRISPR-Cas9 gene editing system and MSC co-culture systems can optimize the transplantation effect of HSCs 109 . In addition, non-viral vector-mediated multi-gene co-transfection technology can enhance the potential of MSCs in treating complex diseases 110 . Furthermore, greater efforts should be made to strengthen the collaborative innovation between basic research and clinical practice, promote the translation between basic research and clinical applications, and establish multi-level policy support systems to facilitate mechanism research, biomarker development, and the optimization of combined treatment strategies—ultimately advancing the clinical application of MSCs in HSCT.
Footnotes
Acknowledgements
The authors thank all collaborators for their valuable contributions to the development of this work.
Ethical Considerations
Not applicable.
Author Contributions
CZ, SM: Conceptualization, Methodology, Investigation, Writing – Original Draft, Writing – Review & Editing. WJ, NM: Data Curation, Formal Analysis. LX: Supervision, Project Administration, Funding, Acquisition. All authors read and approved the final manuscript.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Scientific Research Projects of the Education Department of Anhui Province (2025AHGXZK31294, 2025AHGXZK31426) and Scientific Research Project of Anhui Provincial Health Commission(AHWJ2022a003).
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data Availability Statement
Data sharing is not applicable to this article as no new datasets were generated or analyzed during the current study. Further information can be requested from the corresponding author.
Use of Artificial Intelligence
During the preparation of this work, the authors used Deepseek only for language polishing. No scientific data were generated or modified using AI. The authors take full responsibility for the content of the publication.
Statement of Human and Animal Rights
This study is a review and does not involve human or animal participants.
Statement of Informed Consent
There are no human subjects in this article and informed consent is not applicable.