
Abstract
B-cell lymphoma, the most common subtype of non-Hodgkin lymphoma, presents major therapeutic challenges due to molecular heterogeneity and high relapse rates. While autologous hematopoietic stem cell transplantation (ASCT) has been a cornerstone for relapsed/refractory (R/R) B-cell lymphoma, its efficacy is often compromised by minimal residual disease (MRD) persistence and an immunosuppressive tumor microenvironment. Chimeric antigen receptor (CAR)-T cell therapy has transformed treatment paradigms but faces limited long-term durability due to antigen escape and T-cell exhaustion. The integration of ASCT with CAR-T therapy may offer a complementary approach to address these limitations, leveraging ASCT-induced immune reconstitution to enhance CAR-T-cell persistence and reprogram the tumor milieu. Emerging clinical evidence supports this approach, indicating improved disease control and progression-free survival. Although preliminary clinical outcomes are encouraging, unresolved challenges persist, particularly in terms of cumulative toxicity, optimal therapeutic sequencing, CAR-T-cell longevity, and financial feasibility associated with these advanced therapies. This review provides a comprehensive overview of mechanistic synergies between ASCT and CAR-T therapy, critically evaluates emerging clinical evidence on treatment sequencing, and explores innovative strategies to increase safety, efficacy, and accessibility.

Keywords
Introduction
Lymphoma, a malignant neoplasm of lymphoid origin, remains a major contributor to cancer-related mortality. Non-Hodgkin lymphoma (NHL), the predominant type, comprises a heterogeneous spectrum of B-cell, T-cell, and NK-cell malignancies, with B-cell lymphomas (BCLs) accounting for 80%–90% of cases.1–3 Its pathogenesis is driven by genetic aberrations, dysregulated molecular pathways, and an immunosuppressive tumor microenvironment (TME), all of which contribute to disease progression and treatment resistance.2,4–6 Diffuse large B-cell lymphoma (DLBCL), the most common NHL subtype (30%–40%), exhibits marked molecular heterogeneity.7–11 Its classification into germinal center B-cell-like and activated B-cell-like subtypes highlights distinct oncogenic drivers, with the latter displaying more aggressive clinical behavior and inferior treatment responsiveness. 12
For over 2 decades, R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, prednisone) has been the standard first-line regimen for DLBCL, with cure rates >60%.13–15 However, 10%–15% of patients are refractory, and up to 40% experience relapse, particularly those with MYC/BCL2 translocations, TP53 mutations, or dual-hit signatures.15,16 Recent phase III trials (ZUMA-7, TRANSFORM) have established that CD19-targeted chimeric antigen receptor (CAR)-T cell therapy, rather than autologous hematopoietic stem cell transplantation (ASCT), is now the preferred second-line treatment for patients with early relapse (≤12 months) after frontline therapy.17–19 In this updated paradigm, ASCT is reserved for late-relapse (>12 months) or chemotherapy-sensitive disease, where combining it with novel agents may enhance outcomes. Furthermore, advanced age, frailty, and comorbidities frequently limit ASCT eligibility, underscoring the urgent need for alternative strategies. 20
Such strategies are emerging from advances in precision medicine, which have introduced targeted approaches such as BCL-2 inhibitors, monoclonal antibodies, antibody–drug conjugates, bispecific antibodies, and cellular immunotherapies like CAR-T cells.13,21,22 CAR-T therapy has transformed outcomes in relapsed or refractory (R/R) BCL, particularly in heavily pretreated patients, 23 but challenges remain, including T-cell exhaustion, antigen escape, suboptimal persistence, severe toxicity, and reduced activity in high-risk subtypes.15,24,25
These limitations have prompted interest in ASCT-CAR-T integration, particularly for patients with late relapse (>12 months) or chemotherapy-sensitive disease—a biologically plausible strategy currently under clinical investigation.26,27 By resetting immune homeostasis and creating a supportive environment for CAR-T expansion and persistence, ASCT may help overcome antigen escape, immune exhaustion, and the suppressive TME.28–30 Early clinical evidence, including the ongoing trials NCT05755828 and NCT06381830, suggests that this approach may significantly prolong progression-free survival (PFS) and overall survival (OS), although its magnitude and durability remain to be determined, particularly in high-risk patients.20,31,32
Given the paradigm shift established by recent trials, defining the appropriate patient subsets and clinical scenarios for ASCT-CAR-T integration becomes increasingly essential. This review systematically explores the synergistic mechanisms, clinical challenges, and future directions of CAR-T therapy integrated with ASCT in BCL. Future investigations should focus on optimizing conditioning regimens, refining patient selection, and determining the optimal timing of ASCT relative to CAR-T-cell infusion to maximize therapeutic efficacy. Furthermore, innovative strategies such as genetically engineered stem cell–derived immune cells and metabolic reprogramming of CAR-T cells warrant further exploration to enhance durability and minimize relapse.
ASCT in BCL
ASCT remains a key therapeutic approach in chemotherapy-sensitive R/R BCL, particularly in patients who achieve complete remission (CR) prior to transplantation. 33 Large-cohort studies have demonstrated that ASCT provides durable remission, with 4-year OS and PFS rates reaching 75% and 69%, respectively. 34 Compared with allogeneic hematopoietic stem cell transplantation (allo-HSCT) (Fig. 1), ASCT circumvents the need for a suitable donor, mitigates the risk of graft-versus-host disease (GVHD), and results in a more favorable toxicity profile, thereby expanding its applicability to a broader patient population, particularly younger patients without significant comorbidities and those demonstrating robust chemotherapy sensitivity. 35 In addition, ASCT can influence long-term immune surveillance and post-transplant disease control.28–30 As treatment paradigms evolve, its limitations are increasingly recognized, prompting refinement of transplantation strategies and integration of novel approaches. 36

Comparative mechanisms and clinical outcomes of ASCT and allo-HSCT. Autologous stem cell transplantation (ASCT) is primarily indicated for chemo-sensitive relapsed/refractory patients, involving the reinfusion of autologous hematopoietic stem cells following high-dose chemotherapy to restore hematopoiesis. In contrast, allogeneic hematopoietic stem cell transplantation (allo-HSCT) is reserved for high-risk patients who have failed chemotherapy or ASCT, utilizing stem cells from a human leukocyte antigen-compatible donor to leverage the graft-versus-tumor effect for enhanced disease eradication. Despite their therapeutic potential, both procedures carry significant risks, including infection and relapse. However, ASCT is predominantly associated with mucositis and infection, while allo-HSCT confers an increased risk of graft-versus-host disease. Optimizing transplant strategies and post-transplant interventions remains critical to improving long-term outcomes. Created with BioRender.com.
Relapse remains a major concern, particularly in early recurrence, which is strongly associated with inferior survival outcomes. 37 Even in patients achieving CR, measurable residual disease (MRD) at transplantation critically undermines long-term control, with MRD-positive patients showing a 5-year PFS of ~13%. 38 Residual malignant clones may persist due to impaired immune surveillance during delayed reconstitution and may undergo clonal evolution, leading to immune escape through altered antigen processing, human leukocyte antigen (HLA) class I downregulation, and expansion of resistant subclones. 38 The 5-year relapse rate after ASCT can reach 48.1%, with median OS after relapse of only 0.7 years. 39 Salvage options are limited, and intensified conditioning regimens (e.g., R-BEAM, 90Y-ibritumomab tiuxetan) have not improved long-term survival. 40
Toxicity also limits ASCT, particularly in elderly or heavily pretreated patients. 41 Severe organ toxicity and treatment-related mortality (TRM) remain concerns, and efficacy is reduced in those with multiple prior therapies, failure to achieve CR, or poor performance status. 42 Given these challenges, conventional maintenance strategies (e.g., PD-1 inhibitors, Bruton tyrosine kinase inhibitors) have shown inconsistent benefit in high-risk populations, 43 likely due to post-ASCT immunosuppression and intrinsic tumor resistance (e.g., JAK-STAT alterations). 44 Consequently, next-generation cellular therapies such as CAR-T, capable of MRD eradication, antigen-specific surveillance, and immune reprogramming, are being explored to overcome ASCT’s limitations and improve long-term outcomes. 45
CAR-T therapy in BCL
CAR-T therapy has redefined the treatment paradigm for R/R BCL, resulting in unprecedented response rates and durable remission in selected patients, surpassing conventional salvage chemotherapy. 46 Pivotal trials such as ZUMA-1 and JULIET established CAR-T therapy as a standard option for R/R large B-cell lymphoma (LBCL), 47 leading to regulatory approval of multiple products, including axicabtagene ciloleucel (axi-cel), lisocabtagene maraleucel (liso-cel), and tisagenlecleucel (tisa-cel).25,48
Building upon these foundational achievements in heavily pretreated populations, subsequent clinical efforts have focused on advancing CAR-T therapy into earlier lines of treatment. In addition to its established role in R/R LBCL, CAR-T-cell therapy has now been firmly positioned as the preferred second-line treatment option for patients with early R/R disease, traditionally treated with salvage chemotherapy followed by ASCT. 19 This paradigm shift was driven by two pivotal phase III randomized trials, ZUMA-7 and TRANSFORM, which demonstrated that CD19-targeted CAR-T-cell products (axi-cel and liso-cel) significantly outperformed standard of care (SOC) in this challenging patient population.17–19 In ZUMA-7, axi-cel significantly prolonged event-free survival (EFS) compared with SOC (8.3 vs 2.0 months) and improved complete response (CR) rates (65% vs 32%). 19 Similarly, TRANSFORM reported that liso-cel resulted in superior CR rates (74% vs 43%) and median EFS (not reached vs 2.4 months) relative to SOC. 17 Moreover, patient-reported outcome analyses from ZUMA-7 indicated that CAR-T therapy not only achieved superior disease control but also enhanced global health status, physical functioning, and overall quality of life. 18 Notably, in ZUMA-7, only 36% of patients randomized to the SOC arm were ultimately able to proceed to ASCT, underscoring not only the inferior efficacy but also the limited feasibility of this approach in early relapsed LBCL. 19 These results underscore the superiority of CAR-T therapy over SOC in early relapsed LBCL, firmly establishing it as the new SOC and displacing ASCT in this setting.
While these pivotal trials have established CAR-T therapy as the preferred second-line treatment for early relapsed LBCL, emerging real-world experience and extended follow-up have underscored that durable remissions remain elusive for many patients, and multiple biological and clinical challenges continue to limit the full potential of this transformative modality (Fig. 2) (Table 1). Intrinsic CAR-T-cell dysfunction remains a critical issue. T-cell exhaustion and limited persistence often result in relapse, as prolonged antigen exposure and suppressive TME signals induce functional decline. 49 Persistent stimulation leads to the upregulation of inhibitory receptors such as PD-1, TIM-3, and LAG-3, which suppress T-cell effector functions and cytokine secretion. 50
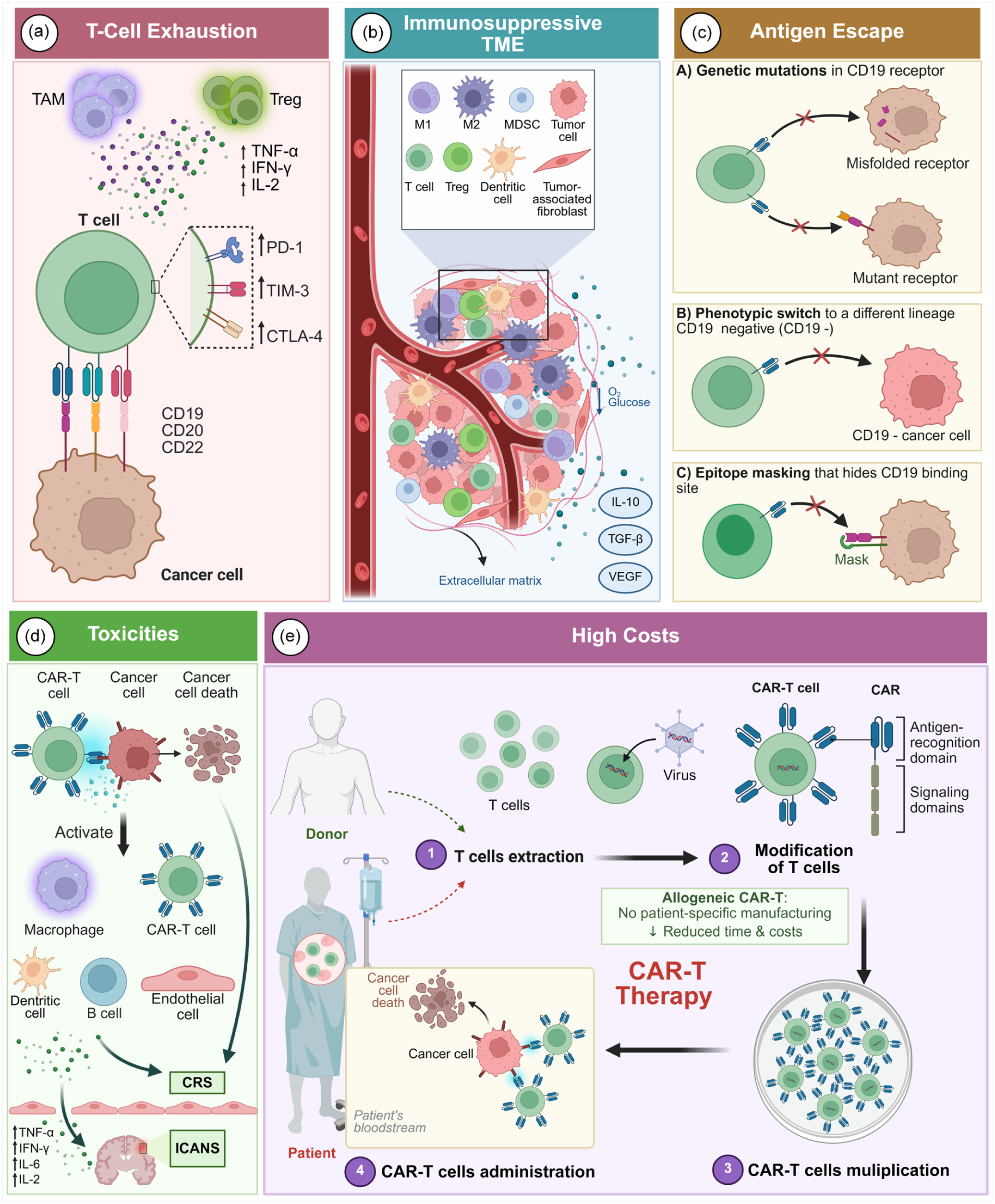
Major limitations of CAR-T therapy in B-cell lymphoma. The efficacy and clinical translation of CAR-T therapy are hindered by multiple barriers, including cellular dysfunction, tumor immune evasion, treatment-related toxicities, and economic challenges. (a) T-cell exhaustion, driven by upregulated inhibitory receptors (e.g., PD-1, TIM-3, CTLA-4) and immunosuppressive cytokines (e.g., IL-10, TGF-β). (b) TME-mediated immune suppression, characterized by the accumulation of Tregs, TAMs, and MDSCs, along with the release of inhibitory cytokines (e.g., IL-10, TGF-β, VEGF). (c) Antigen escape, leading to loss of target antigen due to genetic mutations, lineage switching, or epitope masking. (d) Treatment-related toxicities, including cytokine release syndrome (CRS) and neurotoxicity (ICANS), primarily driven by hyperinflammation and disruption of the blood–brain barrier. (e) High costs, attributed to complex cell engineering, lengthy manufacturing processes, and logistical challenges limiting global accessibility. Cost-reduction strategies, such as the use of allogeneic CAR-T cells, can eliminate patient-specific manufacturing and shorten production time. Created with BioRender.com.
Challenges and optimization strategies for CAR-T for B-cell lymphoma.

PD-1, programmed death-1; PD-L1, programmed death-ligand 1; LAG-3, lymphocyte-activation gene 3; TME, tumor microenvironment; TAMs, tumor-associated macrophages; Tregs, regulatory T cells; MDSCs, myeloid-derived suppressor cells; TGF-β, transforming growth factor-beta; CRS, cytokine release syndrome; CAR, chimeric antigen receptor; ICANS, immune effector cell-associated neurotoxicity syndrome; BBB, blood–brain barrier; CAR-T, chimeric antigen receptor T-cell; BTK, Bruton tyrosine kinase.
Building on the intrinsic dysfunction, tumor-intrinsic escape mechanisms and extrinsic immunosuppressive barriers act in concert to further erode CAR-T-cell efficacy. Antigen escape, particularly CD19 loss, accounts for up to 50% of relapses and is driven by genetic and epigenetic alterations.24,64–66 In addition, the TME, which is enriched with Tregs, myeloid-derived suppressor cells (MDSCs), and inhibitory cytokines such as TGF-β and IL-10, further dampens CAR-T-cell efficacy. 26 Tumor-associated macrophages (TAMs) and stromal cells contribute to metabolic constraints by depleting essential nutrients such as glucose and amino acids, further restricting CAR-T-cell function. 57
Moreover, clinical toxicities and logistical hurdles further limit the widespread application of CAR-T therapy. Severe toxicities, including cytokine release syndrome (CRS) and immune effector cell–associated neurotoxicity syndrome (ICANS), remain significant barriers to broader CAR-T-cell adoption.58,66,79,82 In addition, the high production cost and lengthy manufacturing process (~2–3 weeks) limit accessibility, highlighting the need for novel strategies to streamline production and optimize affordability.74,79
Collectively, these limitations underscore the urgent need for both innovative consolidation strategies and next-generation CAR constructs with improved metabolic fitness, enhanced resistance to immune evasion, and superior persistence to achieve sustained responses in BCL. Among these strategies, ASCT has gained attention as a potentially rational and biologically complementary approach, particularly in selected patients with chemosensitive disease or high-risk features, owing to its ability to reduce tumor burden, reset immune homeostasis, and create a permissive environment for CAR-T expansion and persistence. 30 Integrating ASCT with CAR-T therapy thus represents a potentially valuable avenue for addressing current therapeutic bottlenecks.
Mechanistic synergy of ASCT and CAR-T therapy
CAR-T therapy has revolutionized BCL treatment, yet its long-term durability remains constrained by T-cell exhaustion, antigen escape, and an immunosuppressive TME.26,27 Rather than merely restoring hematopoiesis, ASCT-induced lymphodepletion and hematopoietic recovery reshape the cytokine and metabolic milieu to sustain CAR-T-cell function, fostering immune surveillance enhancement, T-cell receptor (TCR) repertoire diversification, and metabolic reprogramming that may mitigate relapse risk. 30
Following ASCT, hematopoietic recovery initiates profound immune remodeling, replenishing naive and memory T-cell subsets while augmenting IL-7- and IL-15-driven CAR-T-cell expansion.28–30 This cytokine surge reflects homeostatic proliferation triggered by profound lymphodepletion, which activates STAT5 signaling and anti-apoptotic programs (e.g., Bcl-2), preferentially supporting less-differentiated, stem-like CD8⁺ T cells (Tscm)—a subset consistently linked to extended CAR-T-cell persistence and reduced exhaustion.83,84 This lymphodepletion-driven cytokine milieu thus provides a biologic basis for enhanced CAR-T durability post ASCT. Cao et al. 85 reported that 97.1% of patients with ongoing CR exhibited detectable levels of the CD19 CAR transgene 3 months post ASCT, while 68.6% retained detectable CD22 CAR transgene, suggesting a possible role for ASCT in supporting CAR-T-cell persistence. Additionally, ASCT restores dendritic cell function, strengthening antigen presentation and refining CAR-T-cell specificity. 86
Beyond immune reconstitution, TME remodeling through ASCT enhances CAR-T-cell infiltration and metabolic adaptability. 85 The conditioning regimen—high-dose therapy (HDT) and total body irradiation (TBI)—depletes suppressive Tregs and MDSCs, alleviating IL-10- and TGF-β-mediated immune inhibition.30,86–88 This reset reduces nutrient competition, restoring glucose and glutamine availability to CAR-T cells while depriving malignant cells of metabolic advantages.30,88 ASCT-driven mitochondrial rejuvenation has been associated with enhanced oxidative phosphorylation and fatty acid oxidation, which may equip CAR-T cells with sustained bioenergetic capacity.89–91
The integration of ASCT may not only contribute to CAR-T-cell persistence but also enhance MRD clearance and immune surveillance. 92 However, this enhanced surveillance is not absolute; residual tumor cells exploit antigen escape via alternative splicing, epigenetic modifications, and selective clonal expansion.30,93 On one hand, tumor debulking and post-ASCT TCR repertoire diversification may broaden immune recognition and enable detection of antigen-low variants.94–96 On the other hand, the lymphodepletion–reconstitution cycle can impose selective pressure that favors antigen-negative clones through target downregulation or loss, as seen with CD19-negative relapse. 27 On the humoral side, ASCT markedly depletes immunologic memory, including autoreactive and vaccine-induced antibodies, effectively “resetting” B-cell immunity. 97 While this reset may broaden antigen recognition through generation of naïve clones, the transient gap in humoral surveillance during reconstitution could permit clonal expansion of residual malignant cells. 97 Clinically, patients receiving ASCT in combination with CAR-T have shown higher MRD clearance rates and improved PFS (P < 0.01) compared with CAR-T monotherapy, 98 although these findings require further validation.
Furthermore, ASCT cultivates long-lived T-cell clones essential for durable immunosurveillance.83,94,99 By resetting TCR repertoire dynamics, ASCT enables robust antigen recall responses, mitigating the risk of tumor recurrence. 96 This immune reprogramming is particularly relevant in central nervous system (CNS)-involved BCL, where ASCT enhances CAR-T-cell trafficking across the blood–brain barrier, potentially optimizing disease control. 100 Ultimately, the synergy between ASCT and CAR-T therapy orchestrates an immune reset that extends disease remission, providing a scientific rationale for further exploration of this combination in future studies.
Clinical sequencing of ASCT and CAR-T therapy in BCL: current evidence and rationale
ASCT before CAR-T therapy: current evidence and clinical outcomes
A post-ASCT window may allow CAR-T therapy to achieve improved expansion, persistence, and antitumor activity. Recent clinical efforts have explored this approach as a post-ASCT consolidation strategy to eradicate MRD and reinforce immune surveillance, although durability remains to be validated. 101 Supporting this concept, clinical studies have demonstrated the feasibility and suggested the potential efficacy of this sequential approach, particularly in high-risk BCL patients (Table 2). Compared with historical outcomes, the administration of CD19/CD22-targeting CAR-T cells post ASCT has been associated with superior remission rates. 85 In patients with aggressive BCLs, ASCT followed by CD19/CD22 CAR-T therapy achieved a 90.5% overall response rate (ORR) and a 2-year PFS of 83.3% (95% CI: 68.2%–91.7%), demonstrating promising efficacy in R/R aggressive BCL. 85 Additionally, Liu et al. 102 reported a complete response rate (CRR) of 72.0% and a 2-year OS of 68.5% in 25 patients with R/R LBCL who underwent this strategy, with no unexpected toxicities observed at a median follow-up of 27 months. Notably, in central nervous system lymphoma (CNSL), this approach achieved an 81.8% ORR and a CRR of 54.5%, with 1-year PFS and OS rates of 74.59% and 82.5%, respectively. 100 A retrospective analysis further revealed that among patients who achieved CR with ASCT, the 2-year PFS was 66.2%, which was significantly greater than the 47.8% reported in the CAR-T-cell cohort (P < 0.001), and ASCT was also associated with a lower relapse rate (27.8% vs. 48%, P < 0.001), supporting its potential role in prolonging disease control. 101
Clinical study outcomes of HSCT integrated with CAR-T-cell therapy for B-cell lymphoma.
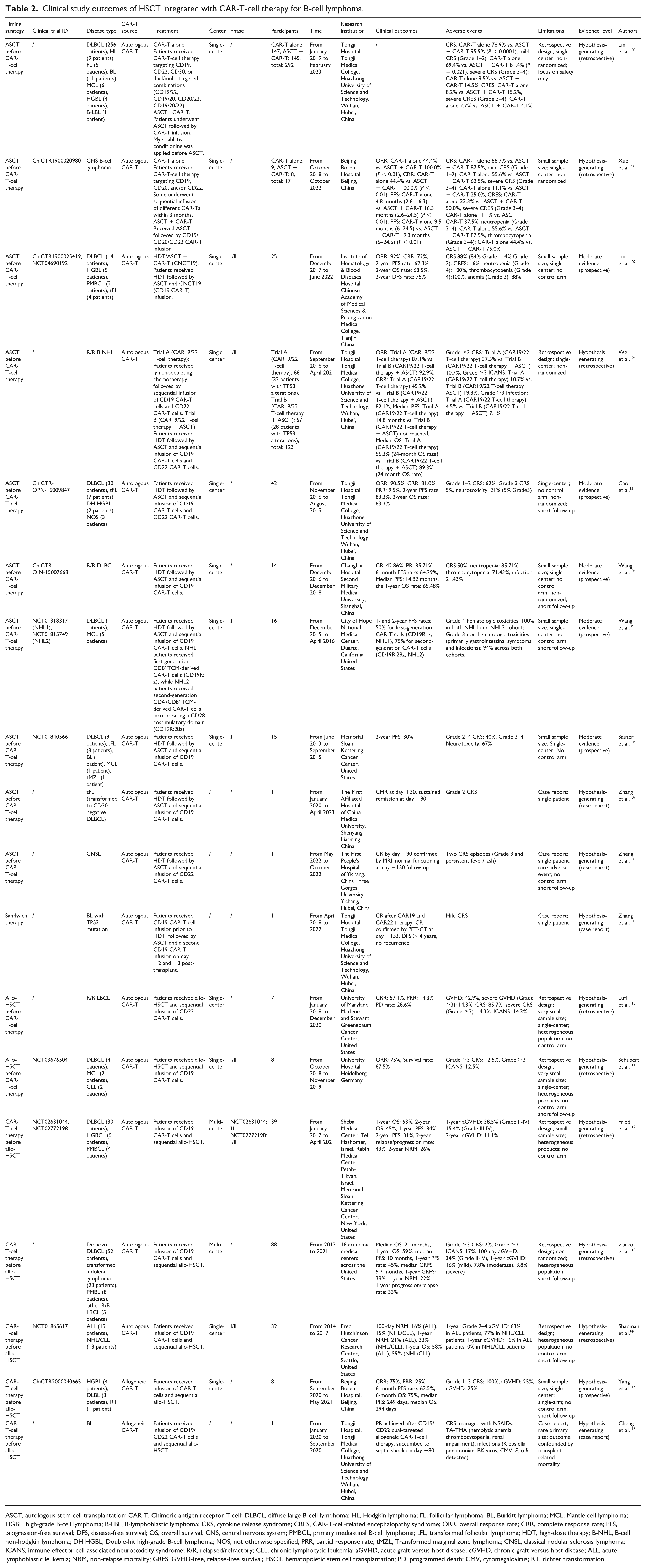
ASCT, autologous stem cell transplantation; CAR-T, Chimeric antigen receptor T cell; DLBCL, diffuse large B-cell lymphoma; HL, Hodgkin lymphoma; FL, follicular lymphoma; BL, Burkitt lymphoma; MCL, Mantle cell lymphoma; HGBL, high-grade B-cell lymphoma; B-LBL, B-lymphoblastic lymphoma; CRS, cytokine release syndrome; CRES, CAR-T-cell-related encephalopathy syndrome; ORR, overall response rate; CRR, complete response rate; PFS, progression-free survival; DFS, disease-free survival; OS, overall survival; CNS, central nervous system; PMBCL, primary mediastinal B-cell lymphoma; tFL, transformed follicular lymphoma; HDT, high-dose therapy; B-NHL, B-cell non-hodgkin lymphoma; DH HGBL, Double-hit high-grade B-cell lymphoma; NOS, not otherwise specified; PRR, partial response rate; tMZL, Transformed marginal zone lymphoma; CNSL, classical nodular sclerosis lymphoma; ICANS, immune effector cell-associated neurotoxicity syndrome; R/R, relapsed/refractory; CLL, chronic lymphocytic leukemia; aGVHD, acute graft-versus-host disease; cGVHD, chronic graft-versus-host disease; ALL, acute lymphoblastic leukemia; NRM, non-relapse mortality; GRFS, GVHD-free, relapse-free survival; HSCT, hematopoietic stem cell transplantation; PD, programmed death; CMV, cytomegalovirus; RT, richter transformation.
This sequential strategy appears particularly beneficial for chemotherapy-sensitive patients at high risk of relapse, including those with persistent MRD or TP53 mutations.104,106 Although ASCT effectively reduces the tumor burden, long-term disease control remains suboptimal when it is used as monotherapy. 106 CAR-T-cell consolidation may provide an opportunity to eliminate residual malignant clones and improve survival outcomes.85,102,105 However, in patients with rapidly progressing disease or resistance to multiple lines of therapy, the prolonged treatment timeline associated with ASCT may not be feasible, making upfront CAR-T therapy a more appropriate option. Thus, precise patient stratification is essential for optimizing the therapeutic synergy between ASCT and CAR-T therapy.
Equally important is determining the optimal infusion timing post ASCT, as hematopoietic reconstitution dynamics may influence CAR-T-cell expansion, persistence, and efficacy. Two primary strategies have been explored: early infusion during the neutropenic phase (before engraftment) and delayed infusion after hematopoietic recovery (Fig. 3a). Early infusion may exploit profound lymphodepletion to enhance CAR-T-cell expansion and tumor clearance but increases infection risk and may delay hematopoietic recovery.30,116 Conversely, delayed infusion may reduce infections and support immune homeostasis but could allow re-emergence of immunoregulatory cells (e.g., Tregs, MDSCs) that suppress CAR-T-cell function and promote antigen escape.30,116 Balancing these trade-offs remains a priority for future studies, particularly in high-risk patients.
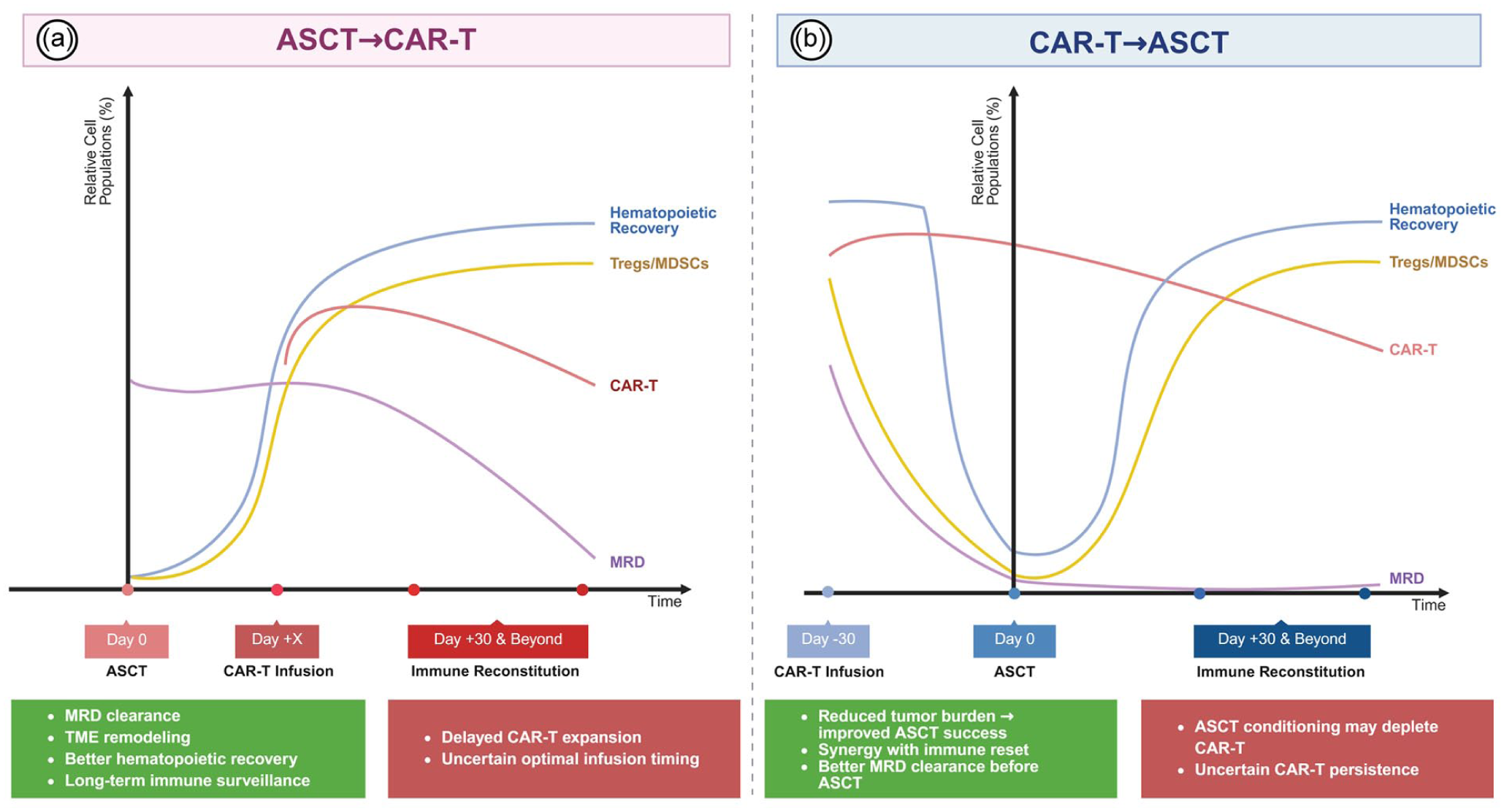
Immune kinetics and clinical implications of ASCT before CAR-T versus CAR-T before ASCT in B-cell lymphoma. ASCT before CAR-T leverages hematopoietic recovery to enhance CAR-T expansion and long-term immune surveillance. CAR-T infusion after ASCT occurs in a lymphodepleted, cytokine-rich environment (e.g., IL-7, IL-15), which can promote robust expansion and optimize MRD clearance but also increases CRS/ICANS risk and infection susceptibility if immune recovery is delayed. In contrast, CAR-T before ASCT can reduce tumor burden, synergize with immune reset, and facilitate ASCT success through improved MRD clearance, but ASCT conditioning may deplete CAR-T cells and limit their persistence. Created with BioRender.com.
CAR-T therapy before transplantation: lessons from allo-HSCT and potential implications for ASCT
Given the critical role of pre-transplant disease burden in determining post-ASCT outcomes, 117 integrating CAR-T therapy before ASCT has emerged as a potential strategy to improve disease control. 99 Evidence from allo-HSCT suggests that CAR-T therapy can serve as a bridge to transplantation by reducing the tumor burden, eradicating MRD, and modulating the immune microenvironment to facilitate hematopoietic engraftment (Table 2) (Fig. 3b).
Retrospective analyses indicate that patients who undergo CAR-T therapy before allo-HSCT exhibit superior long-term survival, particularly when transplantation is performed in a state of CR. A multicenter study reported that among 39 patients receiving allo-HSCT following CAR-T therapy, the 2-year OS reached 45% (95% CI: 31%–66%), with a PFS of 31% (95% CI: 19%–50%). 112 Notably, patients in CR at the time of transplantation demonstrated significantly better OS than those with partial remission or progressive disease did, reinforcing the importance of achieving deep remission before transplantation. 112 A larger cohort study further reported 1-year OS and PFS rates of 59% and 45%, respectively, with a GVHD-free, relapse-free survival (GRFS) of 39%. 113 These findings underscore the critical impact of pre-transplant disease control on long-term survival and suggest that CAR-T therapy before ASCT could potentially confer similar benefits, although this remains to be formally demonstrated.
While current evidence is predominantly derived from allo-HSCT, the underlying principles—disease burden reduction, MRD clearance, and immune modulation—are equally relevant to ASCT. Retrospective studies consistently demonstrate that achieving MRD negativity before ASCT is associated with improved survival. 114 Given the capacity of CAR-T therapy to induce deep remission, its pre-ASCT application may facilitate MRD eradication and potentially reduce post-transplant relapse risk. 114 Additionally, Shadman et al. 99 have reinforced this hypothesis, showing that disease control at the time of transplant remains a key predictor of survival.
Emerging clinical cases further illustrate the feasibility of incorporating CAR-T therapy before ASCT. In a reported case of relapsed TP53-mutated Burkitt lymphoma, a sequential approach involving CAR-T induction, ASCT consolidation, and subsequent CAR-T-cell reinfusion resulted in long-term remission. 109 This “sandwich therapy” highlights the potential synergy between CAR-T therapy and ASCT, which may be particularly relevant for high-risk patients for whom ASCT alone is insufficient; however, this concept requires further validation in prospective studies. 109 Building on these individual reports, the ongoing phase I trial NCT03685786 systematically evaluated the safety and feasibility of ASCT following CAR-T therapy, providing prospective evidence for the feasibility and preliminary efficacy of integrating CAR-T therapy before ASCT in patients with hematologic malignancies. However, its clinical adoption must also consider patient selection, timing, and regimen optimization.
The application of CAR-T therapy before ASCT appears most effective in carefully selected patients.112,113 Those with chemosensitive disease who achieve a meaningful response yet remain at high risk of relapse are more likely to benefit, whereas patients with poor CAR-T-cell expansion, limited persistence, or inadequate disease control after infusion are unlikely to benefit from subsequent ASCT due to residual tumor burden.99,112,113 Data from allo-HSCT indicate that prolonged intervals between CAR-T therapy and transplantation can reduce efficacy. 114 The uncertain durability of CAR-T-cell responses before ASCT raises concerns about relapse before transplantation, and myeloablative conditioning may accelerate CAR-T-cell exhaustion, potentially compromising long-term antitumor activity.
Simultaneous ASCT and CAR-T therapy: a theoretical framework for future exploration
In current BCL management, ASCT and CAR-T therapy are typically administered in sequence. ASCT often serves as a tumor-debulking intervention before CAR-T infusion, whereas CAR-T therapy can function as a bridge to transplantation. A synchronized strategy, in which CAR-T cells are infused during ASCT-induced immune reconstitution, is designed to harness potential immunologic synergy within a single treatment window. ASCT-driven immune remodeling coinciding with ongoing CAR-T–mediated tumor clearance could enhance therapeutic efficacy compared with conventional sequencing. The immune reconstitution phase after ASCT generates a cytokine milieu favorable for CAR-T-cell expansion and persistence, supporting the rationale for synchronized delivery. 30 The clinical feasibility of this approach has not yet been established, and both safety and timing require systematic evaluation in prospective trials.
Challenges in integrating ASCT and CAR-T therapy
The integration of ASCT and CAR-T therapy reshapes the therapeutic landscape for R/R BCL, addressing key limitations of each modality. Despite encouraging early clinical responses, this sequential strategy is hindered by substantial challenges, including compounded toxicities, CAR-T-cell persistence limitations, uncertainty regarding optimal sequencing, and significant economic constraints. Addressing these barriers is imperative for refining therapeutic algorithms and ensuring long-term efficacy.
ASCT-induced immune remodeling exerts paradoxical effects on CAR-T therapy. While the inflammatory surge and lymphodepletion induced by HDT create a transiently favorable niche for CAR-T-cell expansion, these same processes exacerbate toxicity.118,119 Clinical evidence suggests a “dual-phase CRS,” characterized by an early inflammatory surge upon CAR-T-cell infusion, followed by delayed systemic inflammatory activation occurring 5–7 days later. 108 The incidence and severity of CRS are further influenced by ASCT-related systemic inflammation, which amplifies the IL-6 and IL-1 signaling cascades.92,106 Moreover, ASCT may predispose patients to ICANS, particularly through sustained neuroinflammation and disruption of the blood–brain barrier. Notably, in patients with CNSL, ASCT plus CD19/CD22 CAR-T therapy demonstrated manageable toxicity, with no patients with grade ≥3 CRS and only one patient with grade 3 ICANS (7.7%). 100 These findings underscore the intricate interplay between post-transplant inflammation and CAR-T-cell toxicity, highlighting the need for further research into optimal inflammatory modulation strategies.
Hematopoietic suppression remains a major complication, as ASCT-induced myelosuppression prolongs immune recovery and exacerbates CAR-T-cell–associated cytopenias.111,120 A retrospective analysis revealed that severe viral or fungal infections occurred in 22% of patients within 100 days post CAR-T-cell infusion, 99 with cytomegalovirus (CMV) reactivation rates significantly higher in those receiving the integration of ASCT and CAR-T therapy (65.5%) than in those receiving CAR-T-cell monotherapy (28.6%). 121 Additionally, human herpes virus-6 reactivation was observed in 25% of post-ASCT patients, potentially impairing CAR-T cell expansion. 122 The extended duration of hematopoietic recovery also increases the risk of opportunistic infections, further complicating treatment safety. 120
Beyond toxicity management, sustained CAR-T-cell persistence is considered a critical factor potentially contributing to durable remission following ASCT-CAR-T integration. 102 While ASCT effectively reduces the tumor burden and transiently enhances CAR-T-cell expansion by depleting Tregs and MDSCs, these immunosuppressive populations subsequently recover, contributing to CAR-T-cell exhaustion. 88 Moreover, metabolic stress within the TME imposes constraints on CAR-T-cell survival, with increased glucose and glutamine consumption favoring malignant cell persistence. 123 These findings suggest that optimizing CAR-T-cell metabolic fitness and immune surveillance in the post-ASCT setting may be important for improving outcomes.
The optimal timing of CAR-T therapy post ASCT remains an unresolved question with significant therapeutic implications.101,124,125 Early CAR-T-cell administration capitalizes on the post-transplant lymphodepletion phase, enhancing CAR-T-cell expansion, whereas delayed infusion allows for more complete immune reconstitution and reduced toxicity. In a cohort receiving ASCT followed by CD19/CD22 CAR-T therapy, the 2-year PFS reached 83.3% (95% CI: 68.2%–91.7%), suggesting potential benefits associated with this approach. 85 However, in high-risk patients with elevated Center for International Blood and Marrow Transplant Research (CIBMTR) scores, 1-year PFS following CAR-T-cell infusion is significantly lower (34.9%) than that in low-risk patients (75.8%), while the corresponding OS rates are 52.8% and 88.4%, respectively, underscoring the necessity of patient-specific timing strategies. 126 Given the variability in MRD clearance, immune reconstitution kinetics, and TME dynamics, defining an optimal sequencing algorithm remains a critical unmet need.
Economic considerations further complicate the widespread adoption of the integration of ASCT and CAR-T therapy. 127 Although CAR-T therapy typically requires shorter hospitalization durations than ASCT does, its high manufacturing costs and logistical complexity impose significant financial burdens. 93 The integration of ASCT and CAR-T therapy further increases treatment costs due to the dual expenses of cellular therapy and transplantation, along with increased health care resource utilization, which may exacerbate disparities in treatment accessibility. 128 Furthermore, prolonged CAR-T-cell manufacturing poses logistical challenges, particularly in synchronizing production with ASCT-associated hospitalization timelines.
Strategies to optimize the integration of ASCT and CAR-T therapy
Toxicity management
Effective toxicity control is crucial in integrating ASCT and CAR-T therapy. The inflammatory response from ASCT can exacerbate CAR-T-cell-induced CRS, increasing CRS and ICANS risk. 123 Risk stratification before infusion helps identify individuals most susceptible to these overlapping toxicities. High tumor burden, pre-existing neurologic or autoimmune disorders, and post-conditioning elevation of inflammatory biomarkers (CRP, ferritin, IL-6, IL-15) are associated with higher incidence.129–132 The CAR-T construct influences toxicity kinetics: CD28-based products expand more rapidly and trigger earlier peaks, whereas 4-1BB-based products allow partial resolution of transplant-related inflammation before maximal activation. 129 High-risk patients require intensified surveillance, including frequent Immune Effector Cell–Associated Encephalopathy (ICE) score assessments and serial biomarker measurements, to enable intervention before progression to high-grade events. 133
Pharmacologic mitigation of CAR-T-associated toxicities involves targeting IL-6 and IL-1 signaling pathways.134,135 Tocilizumab, the standard intervention for CRS of grade ≥2, has limited penetration across the blood–brain barrier and is therefore unsuitable for isolated ICANS.133,136 In contrast, anakinra crosses the CNS and inhibits IL-1, an upstream mediator of CRS and ICANS.132,133,137 In a retrospective cohort of LBCL, anakinra administered for steroid-refractory or high-risk CRS/ICANS resulted in rapid resolution of neurologic and systemic symptoms without increasing infection rates or disease relapse, supporting its role as a second-line intervention. 138 Building on these observations, a phase II trial (NCT04148430) in 56 recipients of CD28-based products (axi-cel or brexu-cel) showed that prophylactic anakinra, initiated on day −2, reduced the incidence of severe (grade ≥ 3) ICANS to 9.7% compared with historical rates exceeding 30%, without compromising CAR-T delivery or inducing high-grade drug-related toxicity. 139 Most adverse events were mild, and no treatment-related deaths occurred. 139
Engineering-based strategies further enhance safety. Switchable CAR-T-cell constructs and suicide gene systems (e.g., iCasp9) allow real-time modulation of CAR-T-cell activity, providing a potential safeguard against severe toxicity. Additionally, PD-1/CD28 switch receptors have been shown to increase CAR-T-cell tolerance, improve response rates in DLBCL, and reduce neurotoxicity. 53 Similarly, CTLA-4 switch receptors enhance tumor regression while reducing off-target immune toxicity in CD80/86-overexpressing BCL models. 71
Enhancing CAR-T-cell persistence
Long-term CAR-T-cell persistence is essential for sustained remission. While ASCT provides an initial immune clearance window that facilitates CAR-T-cell expansion, prolonged CAR-T-cell survival remains a challenge. To overcome TME-induced immunosuppression, TGF-β/IL-7 chimeric switch receptors convert inhibitory TGF-β signals into IL-7 signals, enhancing CAR-T-cell expansion and anti-tumor activity. 63 Preclinical studies have suggested that this approach may improve CAR-T-cell persistence and tumor control in murine models. 63 Additionally, Bruton’s tyrosine kinase inhibitors, such as ibrutinib, have been shown to augment CAR-T-cell function, mitigate T-cell exhaustion, and modulate the TME by reducing the number of immunosuppressive Tregs and MDSCs. 49
Metabolic reprogramming has also emerged as a promising strategy for enhancing CAR-T-cell persistence. 140 ACAT1 knockdown has been shown to improve CAR-T-cell activation and anti-tumor efficacy by altering cholesterol metabolism. 55 Furthermore, CAR-T cells with enhanced fatty acid oxidation exhibit superior survival in low-glucose environments, with in vivo studies indicating a twofold increase in CAR-T-cell persistence and a reduced risk of T-cell exhaustion. 140 Dual-target CAR-T-cell constructs, including combinations of CD19/CD22, CD19/CD70, and CD19/CD20, have been explored as potential strategies to minimize antigen escape.67,68,73 When combined with PD-1 inhibitors, they synergistically improve long-term remission.59,61
Optimizing treatment sequencing
The timing of CAR-T-cell infusion in the setting of ASCT remains under active investigation, as distinct patient subgroups appear to derive clinical benefit from different sequencing strategies. Infusion approximately 1–2 months after ASCT has been associated with a favorable balance between CAR-T-cell expansion and toxicity risk. 125 In a cohort of patients with R/R aggressive BCL, this approach achieved a 2-year PFS of 83.3% (95% CI: 68.2–91.7). 85 In high-risk individuals, a sequential regimen involving CAR-T infusion before ASCT followed by a second infusion post ASCT has been reported to induce durable remission, as illustrated by a case of R/R Burkitt lymphoma; however, severe CRS occurred, underscoring the need for careful selection. 109 A feasible approach to further optimize sequencing is to incorporate biomarker-driven timing of CAR-T infusion, particularly using MRD and circulating tumor DNA (ctDNA) monitoring before and after ASCT. Integration of such biomarker-guided strategies may refine sequencing decisions. Serial monitoring of MRD by multiparameter flow cytometry or next-generation sequencing, together with ctDNA kinetics and peri-transplant inflammatory markers such as C-reactive protein, ferritin, IL-6, and IL-15, enables dynamic assessment of disease burden and immune status.38,141,142 Persistent MRD or rising ctDNA after ASCT, or biomarker profiles indicating high relapse risk, may warrant earlier CAR-T infusion to prevent overt relapse and control tumor-driven inflammation.38,143 Conversely, MRD-negative patients with pronounced inflammatory activation immediately after ASCT may benefit from delayed infusion until inflammatory parameters normalize, thereby reducing the risk of severe CRS and ICANS. The optimal sequence of ASCT and CAR-T therapy is being investigated in multiple ongoing clinical trials (Table 3) aiming to refine patient-selection criteria and improve long-term outcomes.
Ongoing clinical trials investigating the integration of HSCT and CAR-T therapy in B-cell malignancies.
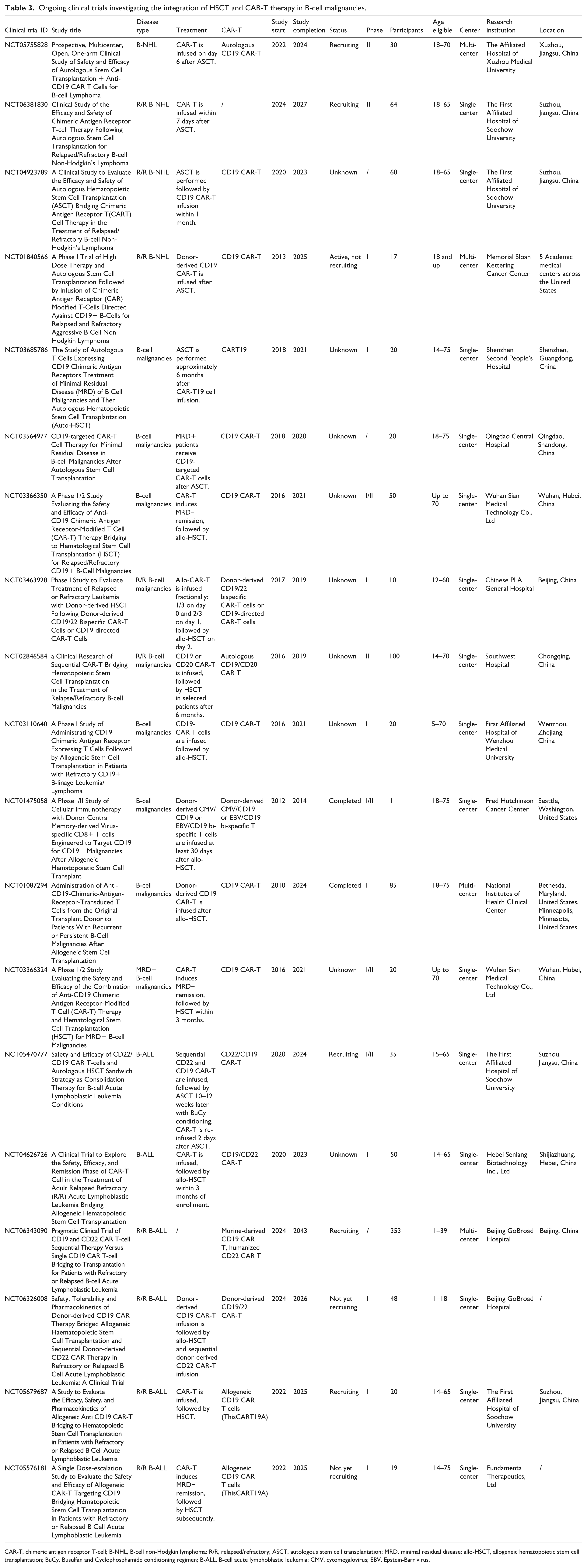
CAR-T, chimeric antigen receptor T-cell; B-NHL, B-cell non-Hodgkin lymphoma; R/R, relapsed/refractory; ASCT, autologous stem cell transplantation; MRD, minimal residual disease; allo-HSCT, allogeneic hematopoietic stem cell transplantation; BuCy, Busulfan and Cyclophosphamide conditioning regimen; B-ALL, B-cell acute lymphoblastic leukemia; CMV, cytomegalovirus; EBV, Epstein-Barr virus.
Economic considerations
The substantial cost of ASCT and CAR-T therapy remains a critical barrier to widespread adoption. 144 CAR-T-cell manufacturing and hospitalization expenses impose a substantial economic burden on patients. Efforts to reduce costs have focused on streamlining CAR-T-cell production, including exploring alternative manufacturing strategies such as mRNA-based CAR-T-cell technology, which has been proposed as a potential means to improve efficiency and cost-effectiveness. 145 Allogeneic (“off-the-shelf”) CAR-T cells offer a complementary strategy by removing the need for patient-specific manufacturing. 146 Donor-derived products are generated in advance, stored, and infused without the 2- to 4-week delay of autologous production, time-saving particularly relevant for patients at high risk of relapse after ASCT.146–148 Early-phase trials of gene-edited allogeneic anti-CD19 CAR-T products, including ALLO-501A, UCART19, and PBCAR0191, have reported ORRs of approximately 50%–67% with low rates of GVHD.147,149 In a recent systematic review and meta-analysis of 334 patients with R/R LBCL, the pooled best ORR was 52.5% (95% CI: 41.0–63.9) with only one reported GVHD event. 148 These results are achieved through targeted disruption of genes essential for host–donor immune recognition.146,149 However, persistence in vivo remains limited, and repeat dosing or additional engineering may be required, potentially offsetting some economic gains.147,148,150 Other approaches to modulate cost include reduced-intensity conditioning ASCT prior to CAR-T infusion, which may preserve efficacy while lowering toxicity and resource use. 151 Additionally, value-based payment models linking reimbursement to clinical outcomes have been proposed to increase affordability and incentivize treatment efficacy.20,152
Conclusion
The integration of ASCT and CAR-T therapy has emerged as a biologically plausible and clinically intriguing approach for R/R BCL, combining the cytoreductive effects of ASCT with the tumor-specific cytotoxicity of CAR-T therapy. By leveraging ASCT-mediated immune reconstitution and CAR-T-driven tumor clearance, this combination enhances MRD eradication, reshapes the TME, and extends disease remission. However, the optimal treatment sequence, long-term CAR-T-cell persistence, and associated toxicities remain key challenges that necessitate further investigation.
Emerging evidence suggests that treatment sequencing significantly influences clinical outcomes. ASCT followed by CAR-T therapy has demonstrated superior MRD clearance and PFS, particularly in high-risk patients with TP53 mutations or PET-PR. Alternatively, CAR-T therapy prior to ASCT, a strategy well-established in allogeneic transplantation, may serve as a bridge to ASCT by reducing the tumor burden and modulating immune homeostasis, thereby improving post-ASCT disease control. Although synchronous administration of ASCT and CAR-T therapy remains largely theoretical, it presents a novel avenue for enhancing CAR-T-cell persistence and immune reconstitution, warranting further exploration.
Despite these advances, it is critical to recognize that the role of ASCT in early-relapsing LBCL has been fundamentally redefined by pivotal trials such as ZUMA-7 and TRANSFORM, which established CAR-T therapy as the new SOC in this setting. Accordingly, the integration of ASCT and CAR-T should be regarded as a strategy tailored for select patient populations, including those with late relapse (>12 months), chemosensitive disease, CNS involvement, or molecular high-risk features.
Additionally, several limitations must be addressed to optimize ASCT-CAR-T-cell integration. The compounded toxicities of ASCT-induced systemic inflammation and CAR-T-cell-mediated immune activation necessitate refined toxicity-management strategies. Additionally, CAR-T-cell persistence remains suboptimal due to metabolic and immunosuppressive constraints within the post-ASCT immune landscape. Economic barriers further challenge the accessibility of this combined approach, underscoring the need for cost-effective strategies such as allogeneic CAR-T-cell development, metabolic reprogramming, and optimized conditioning regimens.
Moving forward, large-scale, multicenter clinical trials will be essential to define optimal sequencing strategies, refine risk-adapted patient-selection criteria, and assess the long-term survival benefits and safety profiles of integrated approaches. Moreover, advances in biomarker-driven immunomonitoring, next-generation CAR constructs (e.g., dual-target, switch receptors), and personalized immune reprogramming are anticipated to enhance therapeutic precision and efficacy. While integrating ASCT with CAR-T therapy offers a biologically plausible and mechanistically synergistic strategy, its clinical value remains to be fully defined. Particularly in high-risk BCL populations, the true clinical impact on long-term outcomes and therapeutic positioning requires rigorous validation through prospective, controlled studies before this approach can be widely adopted.
Footnotes
Ethical Considerations
Not applicable. This review article does not involve any studies with human participants or animals performed by any of the authors.
Statement of Human and Animal Rights
This article does not contain any studies with human or animal subjects.
Statement of Informed Consent
There are no human subjects in this article and informed consent is not applicable.
Author Contributions
XMZ conceptualized and designed the review and drafted the initial manuscript. JYW conducted literature research and prepared figures. YX and XJZ provided supervision and critical intellectual input. All authors contributed to the manuscript and reviewed and approved the final version.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Natural Science Foundation of China (nos. 81873444, 82070213, and 82370196).
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data Availability Statement
Not applicable. This review does not include any datasets generated or analyzed during the current study.